
Simultaneous Quantification of Five Sesquiterpene Components after Ultrasound Extraction in Artemisia annua L. by an Accurate and Rapid UPLC–PDA Assay †

Abstract
:1. Introduction
2. Results
2.1. Single Factor Experiment for Extraction Optimization
2.2. Orthogonal Experiment for Extraction Optimization
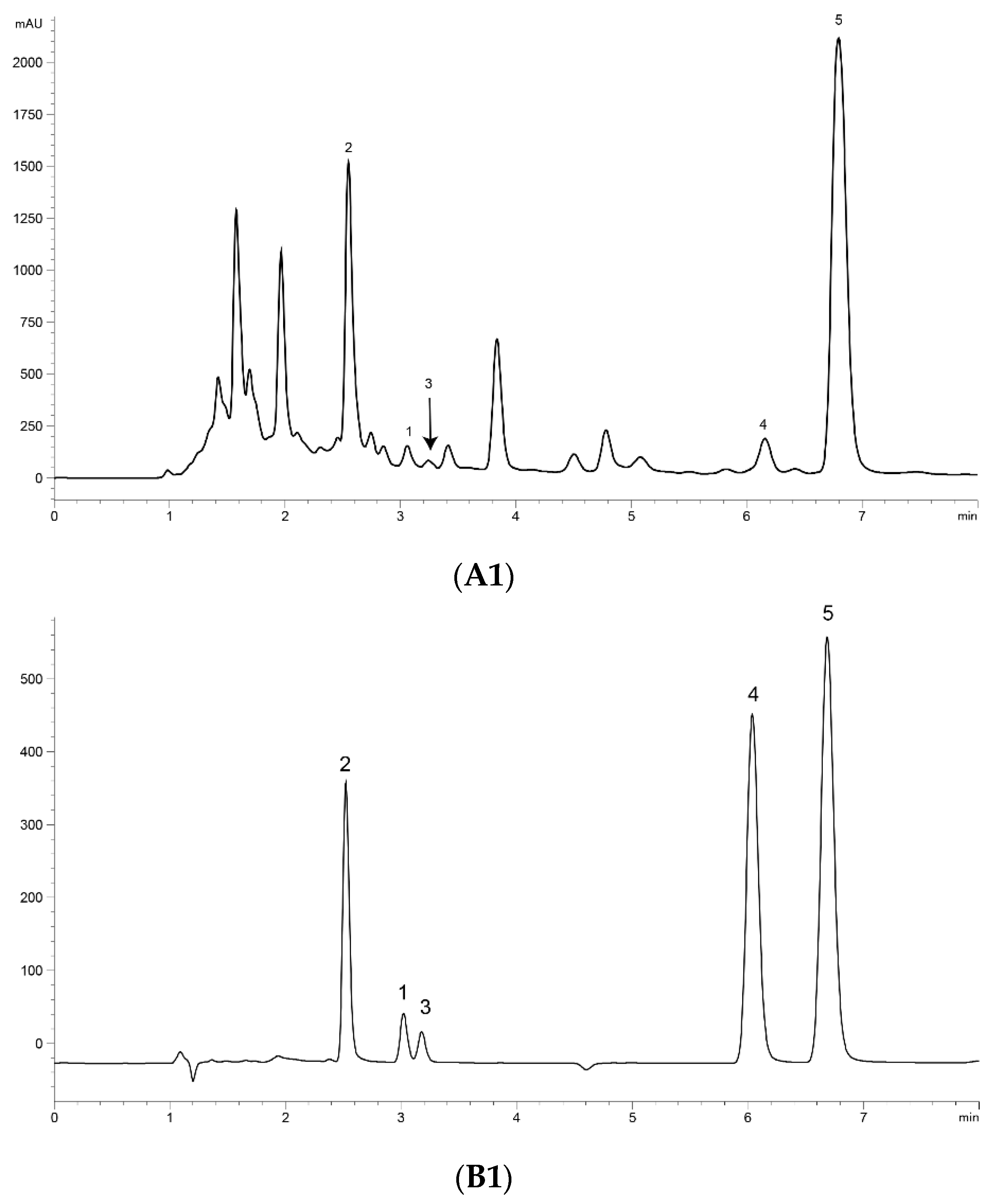
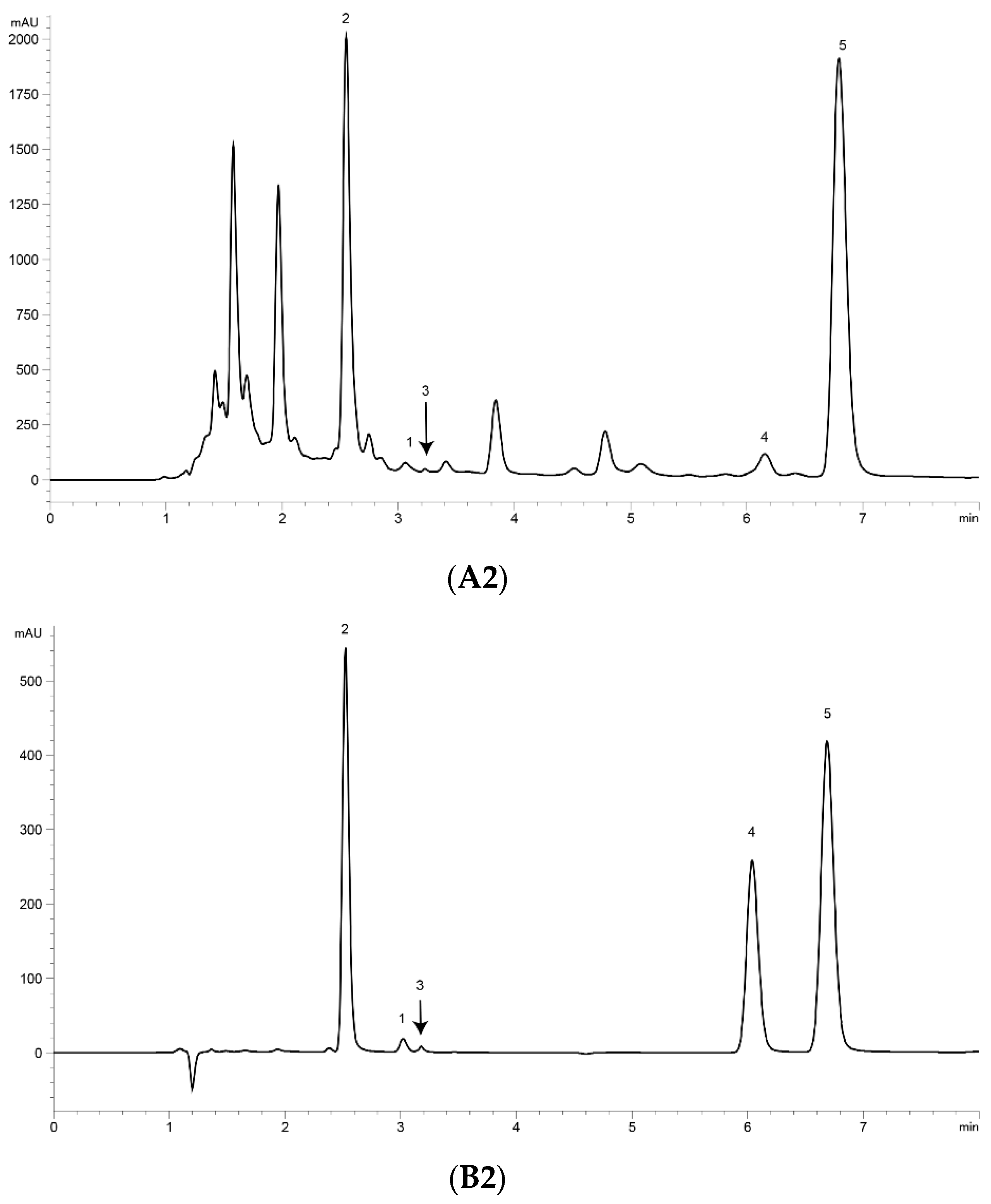
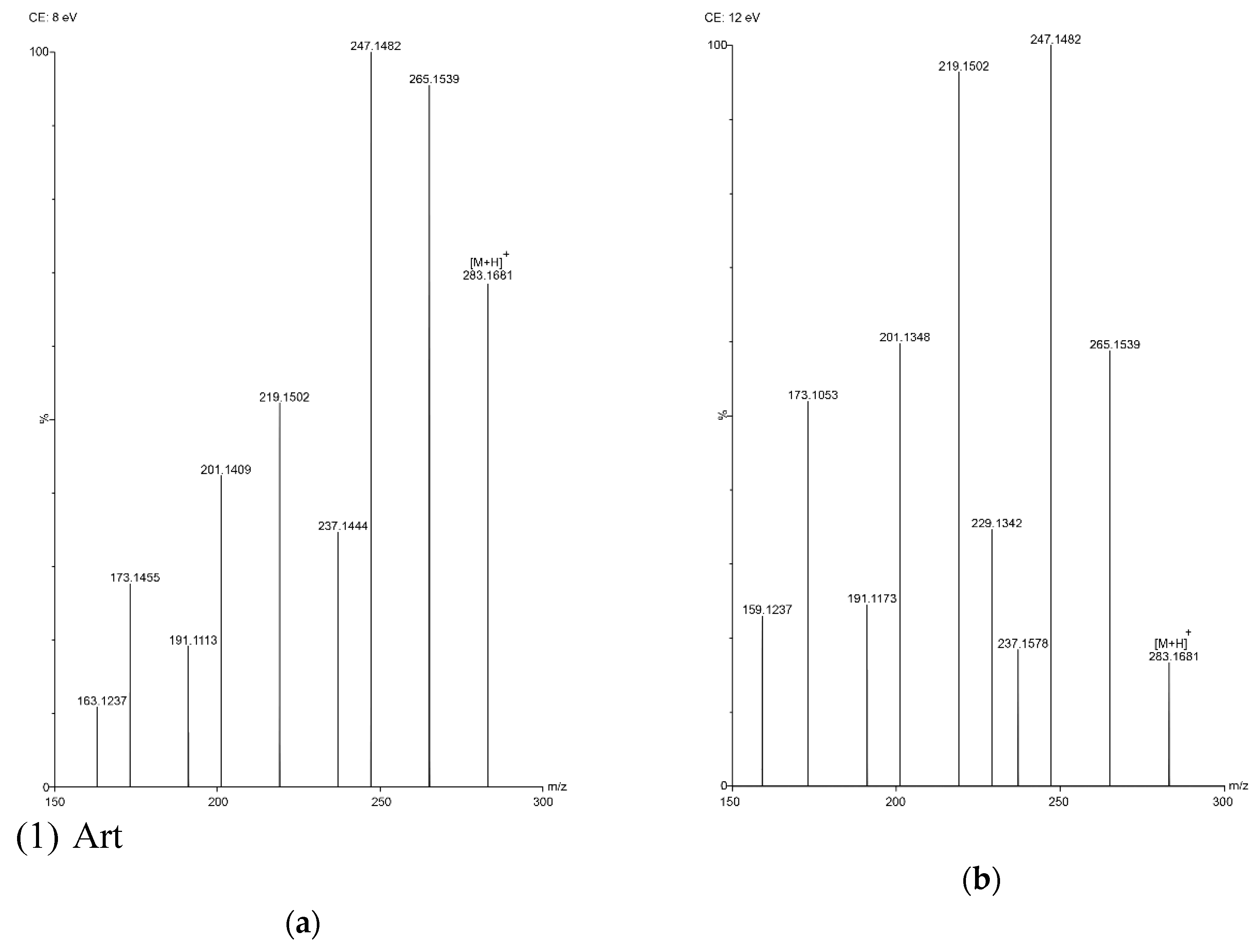
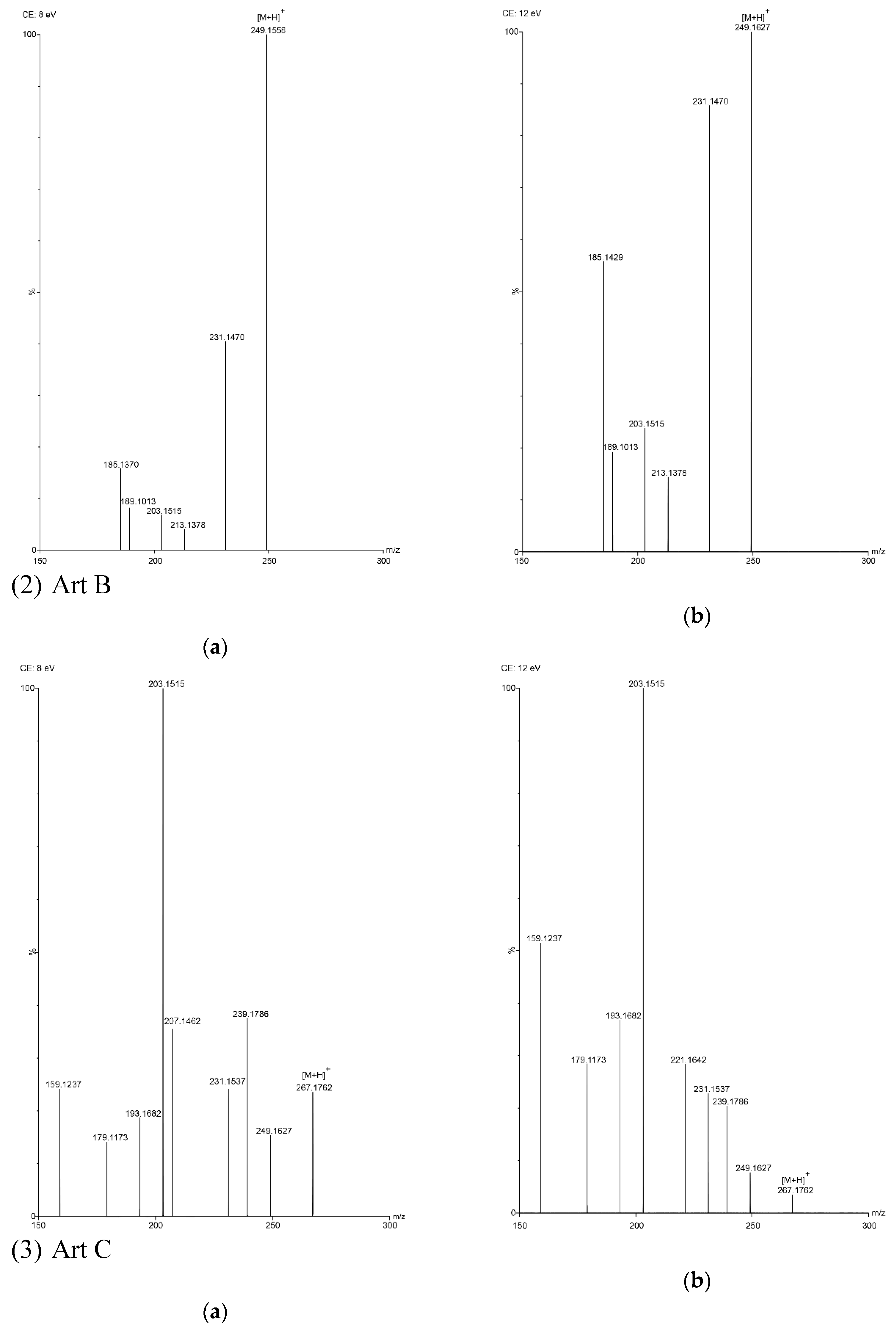
2.3. Optimization of UPLC Separation
2.4. Regression Equation and Linearity
2.5. Precision, Accuracy, Stability, Repeatability and Recovery
2.6. Determination of Content
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Chemicals and Reagents
4.3. Apparatus and Chromatographic Conditions
4.4. Preparation of Standard Solutions
4.5. Preparation of the Extracts and Sample Solutions
4.6. Calibration Curve and Lowest Limit of Quantification (LLOQ)
4.7. Precision, Accuracy, Stability, Repeatability and Recovery
Author Contributions
Funding
Conflicts of Interest
References
- Wang, M.Y. Publication process involving the discovery of artemisinin (qinghaosu) before 1985. Asian Pac. J. Trop. Biomed. 2016, 6, 461–467. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.W.; Tang, W.; Zuo, J.P. Development of artemisinin drugs in the treatment of autoimmune diseases. Sci. Bull. 2016, 61, 37–41. [Google Scholar] [CrossRef]
- Bhakuni, R.S.; Jain, D.C.; Sharma, R.P.; Kumar, S. Secondary metabolites of Artemisia annua and their biological activity. Curr. Sci. 2001, 80, 35–48. [Google Scholar]
- Loo, C.S.N.; Lam, N.S.K.; Yu, D.Y.; Su, X.Z.; Lu, F.L. Artemisinin and its derivatives in treating protozoan infections beyond malaria. Pharm. Res. 2017, 117, 192–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajayi, I.O.; Browne, E.N.; Garshong, B.; Bateganya, F.; Yusuf, B.; Baffour, P.A.; Doamekpor, L.; Balyeku, A.; Munguti, K.; Cousens, S.; et al. Feasibility and acceptability of artemisinin-based combination therapy for the home management of malaria in four African sites. Malar. J. 2008, 7, 1–9. [Google Scholar] [CrossRef]
- Bhandari, P.; Gupta, A.P.; Singh, B.; Kaul, V.K. Simultaneous densitometric determination of artemisinin, artemisinic acid and arteannuin-B in Artemisia annua using reversed-phase thin layer Chromatography. J. Sep. Sci. 2005, 28, 2288–2292. [Google Scholar] [CrossRef]
- Chen, R.R.; Jiang, H.; Pu, H.L. Interaction of artemisinin and its derivatives with human serum albumin studied using spectroscopies and molecular modeling methods. Mol. Biol. Rep. 2013, 40, 4791–4804. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.N.; Chen, C.J.; Li, Q.Q.; Xu, F.R.; Cheng, Y.X.; Dong, X. Monitoring Antifungal Agents of Artemisia annua against Fusarium oxysporum and Fusarium solani, Associated with Panax notoginseng Root-Rot Disease. Molecules 2019, 24, 213. [Google Scholar] [CrossRef] [PubMed]
- Suberu, J.; Gromski, P.S.; Nordon, A.; Lapkin, A. Multivariate data analysis and metabolic profiling of artemisinin andrelated compounds in high yielding varieties of Artemisia annua field-grown in Madagascar. J. Pharm. Biomed. Anal. 2016, 117, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Wu, S.; Lu, X.R.; Zhang, C.; Li, J.; Gong, M.X.; Wang, M.Y. Quality evaluation of the artemisinin-producing plant Artemisia annua L. based on simultaneous quantification of artemisinin and six synergistic components and hierarchical cluster analysis. Ind. Crops Prod. 2018, 118, 131–141. [Google Scholar] [CrossRef]
- Avula, B.; Wang, Y.H.; Smillie, T.J.; Mabusela, W.; Vincent, L.; Weitz, F.; Khan, I.A. Comparison of LC–UV, LC–ELSD and LC–MS methods for the determination of sesquiterpenoids in various species of artemisia. Chromatographia 2009, 70, 797–803. [Google Scholar] [CrossRef]
- Carbonara, T.; Pascale, R.; Argentieri, M.P.; Papadia, P.; Fanizzi, F.P.; Villanova, L.; Avato, P. Phytochemical analysis of a herbal tea from Artemisia annua L. J. Pharm. Biomed. Anal. 2012, 62, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.F.; Gonzalez, J.M. Analysis of underivatized artemisinin and related sesquiterpene lactones by high-performance liquid chromatography with ultraviolet detection. Phytochem. Anal. 2009, 20, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.F.; Wang, H.H.; Lu, X.; Xu, G.W.; Liu, B.Y. Metabolic fingerprinting investigation of Artemisia annua L. in different stages of development by gas chromatography and gas chromatography-mass spectrometry. J. Chromatogr. A 2008, 1186, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.F. Nutrient Deficiency in the Production of Artemisinin, Dihydroartemisinic Acid, and Artemisinic Acid in Artemisia annua L. J. Agric. Food Chem. 2007, 55, 1686. [Google Scholar] [CrossRef]
- Bilia, A.R.; Malgalhaes, P.M.; Bergonzi, M.C.; Vincieri, F.F. Simultaneous analysis of artemisinin and flavonoids of several extracts of Artemisia annua L. obtained from a commercial sample and a selected cultivar. Phytomedicine 2006, 13, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.Q.; Chen, H.L.; Fan, L.Y.; Chen, X.G.; Hu, Z.D. On-line conversion and determination of artemisinin and its kinetic parameters using orthogonal design by coupling of flow injection with capillary electrophoresis. Anal. Chim. Acta 2004, 525, 239–245. [Google Scholar] [CrossRef]
- Jessing, K.K.; Juhler, R.K.; Strobel, B.W. Monitoring of Artemisinin, Dihydroartemisinin, and Artemether in Environmental Matrices Using High-Performance Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS). J. Agric. Food. Chem. 2011, 59, 11735–11743. [Google Scholar] [CrossRef]
- Lapkina, A.A.; Walkerb, A.; Sullivanc, N.; Khambayd, B.; Mlamboa, B.; Chemata, S. Development of HPLC analytical protocols for quantification of artemisinin in biomass and extracts. J. Pharm. Biomed. Anal. 2009, 49, 908–915. [Google Scholar] [CrossRef]
- Chemat, S.; Aissa, A.; Boumechhour, A.; Arous, O.; Amar, H.A. Extraction mechanism of ultrasound assisted extraction and its effect on higher yielding and purity of artemisinin crystals from Artemisia annua L. leaves. Ultrason. Sonochem. 2017, 34, 310–316. [Google Scholar] [CrossRef]
- Graves, R.A.; Ledet, G.A.; Nation, C.A.; Showers, P.R.; Pramar, Y.V.; Mandal, T.K.; Bostanian, L.A. An Ultra High Pressure Liquid Chromatographic Method for the Determination of Artemisinin. Drug Dev. Ind. Pharm. 2015, 41, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Gong, M.X.; Qiu, F.; Li, J.; Wang, M.Y. Effects of arteannuin B, arteannuic acid and scopoletin on pharmacokinetics of artemisinin in mice. Asian Pac. J. Trop. Med. 2016, 9, 677–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, C.; Gong, M.X.; Wang, M.Y. Combination of artemisinin-based natural compounds from Artemisia annua L. for the treatment of malaria: Pharmacodynamic and pharmacokinetic studies. Phytother. Res. 2018, 32, 1415–1420. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.Y.; Zhang, Y.R.; Ji, J.B.; Xing, J. Investigation of the component in Artemisia annua L. leading to enhanced antiplasmodial potency of artemisinin via regulation of its metabolism. J. Ethnopharmacol. 2017, 207, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, Q.; Wu, J.; Wang, M.Y.; Yu, J.X. Artemisinin and Its Derivatives as a Repurposing Anticancer Agent: What Else Do We Need to Do? Molecules 2016, 21, 1331. [Google Scholar] [CrossRef] [PubMed]
- FDA. Guidance for Industry Bioanalytical Method Validation; Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM): Washington, DC, USA, 2001; p. 6.
- Wang, Z.B.; Zhu, W.B.; Liu, H.; Wu, G.S.; Song, M.M.; Yang, B.Y.; Yang, D.Q.; Wang, Q.H.; Kuang, H.X. Simultaneous Determination of Aesculin, Aesculetin, Fraxetin, Fraxin and Polydatin in Beagle Dog Plasma by UPLC-ESI-MS/MS and Its Application in a Pharmacokinetic Study after Oral Administration Extracts of Ledum palustre L. Molecules 2018, 23, 2285. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Linear Range (μg/mL) | Regression Equation (y = ax + b) a) | R | LLOQ b) (μg/mL) | |
|---|---|---|---|---|---|
| Slope (a) | Intercept (b) | ||||
| Art | 31.44–1572 | 0.8332 | 7.996 | 1.000 | 31.44 |
| Art B | 25.48–1274 | 8.265 | 109.9 | 0.9996 | 25.48 |
| Art C | 40.56–2028 | 0.4145 | 1.707 | 0.9998 | 40.56 |
| DHAA | 31.44–1572 | 9.186 | 38.13 | 0.9998 | 31.44 |
| AA | 26.88–1396 | 14.28 | 175.6 | 0.9990 | 26.88 |
| Compound | Fortified Conc. (μg/mL) | Intra-Day (n = 5) | Inter-Day (n = 5) | ||||
|---|---|---|---|---|---|---|---|
| Observed Conc. (μg/mL) | Precision a) (%) | Accuracy b) (%) | Observed Conc. (μg/mL) | Precision (%) | Accuracy (%) | ||
| Art | 786.0 | 817.6 | 0.21 | 104.0 | 807.9 | 1.27 | 102.8 |
| 314.4 | 330.1 | 0.15 | 105.0 | 324.3 | 2.42 | 103.1 | |
| 157.2 | 163.6 | 0.09 | 104.1 | 162.4 | 1.89 | 103.3 | |
| Art B | 637.0 | 660.3 | 0.13 | 103.7 | 605.8 | 0.32 | 95.10 |
| 254.8 | 266.5 | 0.22 | 104.6 | 246.9 | 0.24 | 96.90 | |
| 127.4 | 129.8 | 0.10 | 101.9 | 123.0 | 1.12 | 96.60 | |
| Art C | 1014 | 1068 | 0.63 | 105.3 | 1037 | 2.16 | 102.2 |
| 405.6 | 391.6 | 1.49 | 96.60 | 395.6 | 2.66 | 97.50 | |
| 202.8 | 212.9 | 0.18 | 105.0 | 197.4 | 2.88 | 97.30 | |
| DHAA | 786.0 | 821.4 | 0.08 | 104.5 | 821.4 | 1.69 | 104.5 |
| 314.4 | 327.3 | 0.17 | 104.1 | 321.9 | 2.34 | 102.4 | |
| 157.2 | 161.7 | 0.15 | 102.9 | 158.3 | 2.85 | 100.7 | |
| AA | 672.0 | 701.9 | 0.15 | 104.4 | 705.6 | 2.76 | 105.0 |
| 268.8 | 281.7 | 0.19 | 104.8 | 280.8 | 2.10 | 104.5 | |
| 134.4 | 136.0 | 0.24 | 101.2 | 130.3 | 2.37 | 97.00 | |
| Compound | Stability | Repeatability | RSD (%) |
|---|---|---|---|
| RSD (%) | Retention Time (min) | Peak Area (AU) | |
| Art | 1.63 | 0.069 | 2.37 |
| Art B | 1.65 | 0.060 | 2.42 |
| Art C | 1.52 | 0.070 | 2.04 |
| DHAA | 1.45 | 0.091 | 2.15 |
| AA | 0.96 | 0.092 | 1.64 |
| Compound | Original Conc. | Spiked Conc. | Found Conc. | Recovery a) ± SD | RSD (%) |
|---|---|---|---|---|---|
| (μg/mL) | (μg/mL) | (μg/mL) | (%) | ||
| Art | 116.1 | 117.0 | 233.7 | 100.6 ± 1.28 | 1.27 |
| Art B | 179.5 | 180.0 | 358.8 | 99.6 ± 0.83 | 0.84 |
| Art C | 10.70 | 13.00 | 23.23 | 96.5 ± 2.72 | 2.82 |
| DHAA | 32.90 | 33.00 | 65.70 | 99.4 ± 2.67 | 1.69 |
| AA | 282.5 | 285.0 | 561.6 | 97.9 ± 2.50 | 2.56 |
| No. | Region | Art Content (mg/g) | Art B Content (mg/g) | Art C Content (mg/g) | DHAA Content (mg/g) | AA Content (mg/g) |
|---|---|---|---|---|---|---|
| 1 | Xinjiang | 10.16 ± 0.011 | 5.347 ± 0.0023 | 2.700 ± 0.0025 | - | 3.449 ± 0.0015 |
| 2 | Hubei | 12.04 ± 0.017 | 8.108 ± 0.0015 | 2.228 ± 0.0021 | 0.8521 ± 0.00025 | 15.17 ± 0.0072 |
| 3 | Zhejiang1 | 5.457 ± 0.0031 | 4.581 ± 0.0020 | 1.153 ± 0.0016 | - | 6.459 ± 0.0016 |
| 4 | Shandong1 | 8.604 ± 0.0022 | 8.356 ± 0.0025 | 0.9970 ± 0.00036 | 0.6356 ± 0.00030 | 7.735 ± 0.0021 |
| 5 | Zhejiang2 | 27.59 ± 0.015 | 4.462 ± 0.0015 | 0.9026 ± 0.00021 | - | 3.022 ± 0.0019 |
| 6 | Hebei | 88.79 ± 0.010 | 1.289 ± 0.0020 | - | - | 1.406 ± 0.0021 |
| 7 | Shaanxi | 7.594 ± 0.0021 | 8.593 ± 0.0011 | 1.224 ± 0.0025 | - | 7.620 ± 0.0021 |
| 8 | Guizhou | 13.78 ± 0.010 | 3.332 ± 0.0015 | 1.980 ± 0.0019 | - | 4.499 ± 0.0020 |
| 9 | Hunan1 | 8.216 ± 0.0012 | 0.4597 ± 0.00070 | 0.9990 ± 0.00022 | 2.020 ± 0.0025 | 0.4449 ± 0.00024 |
| 10 | Beijing1 | 5.562 ± 0.0010 | 6.877 ± 0.0020 | 1.379 ± 0.0022 | 3.737 ± 0.0022 | 8.313 ± 0.0013 |
| 11 | Shandong2 | 4.457 ± 0.0020 | 7.391 ± 0.0014 | 1.494 ± 0.0016 | 0.4174 ± 0.00020 | 11.14 ± 0.0034 |
| 12 | Beijing2 | 2.303 ± 0.0019 | 3.562 ± 0.0021 | 0.2121 ± 0.00010 | 0.6527 ± 0.0019 | 5.606 ± 0.0024 |
| 13 | Hunan2 | 6.880 ± 0.0026 | 0.5579 ± 0.00041 | 0.8605 ± 0.00045 | 3.587 ± 0.0019 | 0.5311 ± 0.0078 |
| 14 | Liaoning | 3.649 ± 0.0016 | 6.323 ± 0.0022 | 1.338 ± 0.0017 | - | 6.477 ± 0.0029 |
| 15 | Hunan3 | 63.74 ± 0.010 | 4.028 ± 0.0015 | 2.967 ± 0.0015 | 13.58 ± 0.0057 | 3.231 ± 0.0021 |
| Level | Factors | ||||
|---|---|---|---|---|---|
| A Method | B Solvent | C Ratio (g/mL) | D Time/h | E Cycles | |
| 1 | ultrasonic | methanol | 1:20 | 0.5 | 1 |
| 2 | Condensation reflux | petroleum ether | 1:30 | 1 | 2 |
| 3 | - | ethyl acetate | 1:40 | 2 | 3 |
| Level | Factors | ||
|---|---|---|---|
| X ratio/(g/mL) | Y time/h | Z times | |
| a | 1:10 | 0.25 | 1 |
| b | 1:20 | 0.5 | 2 |
| c | 1:30 | 1 | 3 |
| No. | X | Y | Z |
|---|---|---|---|
| 1 | a | a | a |
| 2 | a | b | b |
| 3 | a | c | c |
| 4 | b | a | b |
| 5 | b | b | c |
| 6 | b | c | a |
| 7 | c | a | c |
| 8 | c | b | a |
| 9 | c | c | b |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruan, J.; Liu, Z.; Qiu, F.; Shi, H.; Wang, M. Simultaneous Quantification of Five Sesquiterpene Components after Ultrasound Extraction in Artemisia annua L. by an Accurate and Rapid UPLC–PDA Assay. Molecules 2019, 24, 1530. https://doi.org/10.3390/molecules24081530
Ruan J, Liu Z, Qiu F, Shi H, Wang M. Simultaneous Quantification of Five Sesquiterpene Components after Ultrasound Extraction in Artemisia annua L. by an Accurate and Rapid UPLC–PDA Assay. Molecules. 2019; 24(8):1530. https://doi.org/10.3390/molecules24081530
Chicago/Turabian StyleRuan, Jiaqi, Zhengyue Liu, Feng Qiu, Henan Shi, and Manyuan Wang. 2019. "Simultaneous Quantification of Five Sesquiterpene Components after Ultrasound Extraction in Artemisia annua L. by an Accurate and Rapid UPLC–PDA Assay" Molecules 24, no. 8: 1530. https://doi.org/10.3390/molecules24081530