
In Vitro and In Silico Acetylcholinesterase Inhibitory Activity of Thalictricavine and Canadine and Their Predicted Penetration across the Blood-Brain Barrier

 , , , , , , , and
, , , , , , , and
Abstract
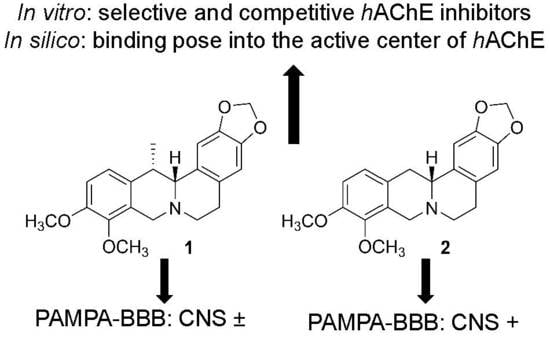
:
1. Introduction
2. Results and Discussion
2.1. AChE and BChE Inhibition Studies
2.2. AChE Kinetic Studies
2.3. Docking Studies
2.4. BBB Permeability
3. Materials and Methods
3.1. Instruments
3.2. Materials and Chemicals
3.3. AChE and BChE Inhibition
3.4. Kinetic Study of AChE Inhibition
3.5. Molecular Docking Studies
3.6. Prediction of CNS Permeability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lleó, A. Current Therapeutic Options for Alzheimer’s Disease. Curr. Genom. 2007, 8, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y. Potential therapeutic agents against Alzheimer’s disease from natural sources. Arch. Pharm. Res. 2010, 33, 1589–1609. [Google Scholar] [CrossRef] [PubMed]
- Rasool, M.; Malik, A.; Qureshi, M.S.; Manan, A.; Pushparaj, P.N.; Asif, M.; Qazi, M.H.; Qazi, A.M.; Kamal, M.A.; Gan, S.H.; Sheikh, I.A. Recent updates in the treatment of neurodegenerative disorders using natural compounds. Evid. Based Complement. Alternat. Med. 2014, 2014, 979730. [Google Scholar] [CrossRef]
- Lahiri, D.K.; Farlow, M.R.; Greig, N.H.; Sambamurti, K. Current drug targets for Alzheimer’s disease treatment. Drug Dev. Res. 2002, 56, 267–281. [Google Scholar] [CrossRef]
- Nordberg, A.; Ballard, C.; Bullock, R.; Darreh-Shori, T.; Somogyi, M. A Review of butyrylcholinesterase as a therapeutic target in the treatment of Alzheimer’s Disease. Prim. Care Companion CNS Disord. 2013, 15, 12r01412. [Google Scholar] [CrossRef]
- Ehret, M.J.; Chamberlin, K.W. Current practices in the treatment of Alzheimer disease: Where is the evidence after the Phase III trials? Clin. Ther. 2015, 37, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Gill, K.D.; Mahdi, A.A. Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology 2014, 76, 27–50. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.S.; Mangialasche, F.; Andreasen, N.; Feldman, H.; Giacobini, E.; Jones, R.; Mantua, V.; Mecocci, P.; Pani, L.; Winblad, B.; et al. Clinical trials and late-stage drug development for Alzheimer’s disease: an appraisal from 1984 to 2014. J. Intern. Med. 2014, 275, 251–283. [Google Scholar] [CrossRef] [Green Version]
- Zemek, F.; Drtinova, L.; Nepovimova, E.; Sepsova, V.; Korabecny, J.; Klimes, J.; Kuca, K. Outcomes of Alzheimer’s disease therapy with acetylcholinesterase inhibitors and memantine. Expert Opin. Drug Saf. 2014, 13, 759–774. [Google Scholar] [PubMed]
- Giacobini, E. Cholinesterase inhibitors: new roles and therapeutic alternatives. Pharmacol. Res. 2004, 50, 433–440. [Google Scholar] [CrossRef]
- Kandiah, N.; Pai, M.-C.; Senanarong, V.; Looi, I.; Ampil, E.; Park, K.W.; Karanam, A.K.; Christopher, S. Rivastigmine: the advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia. Clin. Interv. Aging 2017, 12, 697–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H. New insights into huperzine A for the treatment of Alzheimer’s disease. Acta Pharmacol. Sin. 2012, 33, 1170–1175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-M.; Hu, G.-Y. Huperzine A, a nootropic alkaloid, inhibits N-methyl-D-aspartate-induced current in rat dissociated hippocampal neurons. Neuroscience 2001, 105, 663–669. [Google Scholar] [CrossRef]
- Hostettmann, K.; Borloz, A.; Urbain, A.; Marston, A. Natural product inhibitors of acetylcholinesterase. Curr. Org. Chem. 2006, 10, 825–847. [Google Scholar] [CrossRef]
- Mukherjee, P.K.; Kumar, V.; Mal, M.; Houghton, P.J. Acetylcholinesterase inhibitors from plants. Phytomedicine 2007, 14, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Houghton, P.J.; Ren, Y.; Howes, M.-J. Acetylcholinesterase inhibitors from plants and fungi. Nat. Prod. Rep. 2006, 23, 181–199. [Google Scholar] [CrossRef]
- Chlebek, J.; Macáková, K.; Cahlíkovi, L.; Kurfürst, M.; Kunes, J.; Opletal, L. Acetylcholinesterase and butyrylcholinesterase inhibitory compounds from Corydalis cava (Fumariaceae). Nat. Prod. Commun. 2011, 6, 607–610. [Google Scholar] [CrossRef]
- Adsersen, A.; Kjølbye, A.; Dall, O.; Jäger, A.K. Acetylcholinesterase and butyrylcholinesterase inhibitory compounds from Corydalis cava Schweigg. & Kort. J. Ethnopharmacol. 2007, 113, 179–182. [Google Scholar]
- Adsersen, A.; Gauguin, B.; Gudiksen, L.; Jäger, A.K. Screening of plants used in Danish folk medicine to treat memory dysfunction for acetylcholinesterase inhibitory activity. J. Ethnopharmacol. 2006, 104, 418–422. [Google Scholar] [CrossRef]
- Zeng, H.; Wu, X. Alzheimer’s disease drug development based on computer-aided drug design. Eur. J. Med. Chem. 2016, 121, 851–863. [Google Scholar] [CrossRef]
- Bermudez-Lugo, J.A.; Rosales-Hernandez, M.C.; Deeb, O.; Trujillo-Ferrara, J.; Correa-Basurto, J. In silico methods to assist drug developers in acetylcholinesterase inhibitor design. Curr. Med. Chem. 2011, 18, 1122–1136. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, J.E.; Pigni, N.B.; Andujar, S.A.; Roitman, G.; Suvire, F.D.; Enriz, R.D.; Tapia, A.; Bastida, J.; Feresin, G.E. Alkaloids from Hippeastrum argentinum and their cholinesterase-inhibitory activities: An in vitro and in silico study. J. Nat. Prod. 2016, 79, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Ordóñez, W.O.; Tamarozzi, E.R.; da Silva, G.M.; Aristizabal-Pachón, A.F.; Sakamoto-Hojo, E.T.; Takahashi, C.S.; Giuliatti, S. Exploration of the acetylcholinesterase inhibitory activity of some alkaloids from Amaryllidaceae family by molecular docking in silico. Neurochem. Res. 2017, 42, 2826–2830. [Google Scholar] [CrossRef] [PubMed]
- Cortes, N.; Alvarez, R.; Osorio, E.H.; Alzate, F.; Berkov, S.; Osorio, E. Alkaloid metabolite profiles by GC/MS and acetylcholinesterase inhibitory activities with binding-mode predictions of five Amaryllidaceae plants. J. Pharm. Biomed. Anal. 2015, 102, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Adhami, H.R.; Linder, T.; Kaehlig, H.; Schuster, D.; Zehl, M.; Krenn, L. Catechol alkenyls from Semecarpus anacardium: Acetylcholinesterase inhibition and binding mode predictions. J. Ethnopharmacol. 2012, 139, 142–148. [Google Scholar] [CrossRef] [PubMed]
- da Silva, V.B.; de Andrade, P.; Kawano, D.F.; Morais, P.A.B.; de Almeida, J.R.; Carvalho, I.; Taft, C.A.; da Silva, C.H.T.D.P. In silico design and search for acetylcholinesterase inhibitors in Alzheimer’s disease with a suitable pharmacokinetic profile and low toxicity. Future Med. Chem. 2011, 3, 947–960. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Crossing the blood-brain barrier: are we getting it right? Drug Discov. Today 2001, 6, 1–2. [Google Scholar] [CrossRef]
- Abbott, N.J. Blood-brain barrier structure and function and the challenges for CNS drug delivery. J. Inherit. Metab. Dis. 2013, 36, 437–449. [Google Scholar] [CrossRef]
- van Asperen, J.; Mayer, U.; van Tellingen, O.; Beijnen, J.H. The functional role of P-glycoprotein in the blood-brain barrier. J. Pharm. Sci. 1997, 86, 881–884. [Google Scholar] [CrossRef]
- Nielsen, P.A.; Andersson, O.; Hansen, S.H.; Simonsen, K.B.; Andersson, G. Models for predicting blood-brain barrier permeation. Drug Discov. Today 2011, 16, 472–475. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Cahlíková, L.; Hulová, L.; Hrabinová, M.; Chlebek, J.; Hošťálková, A.; Adamcová, M.; Šafratová, M.; Jun, D.; Opletal, L.; Ločárek, M.; et al. Isoquinoline alkaloids as prolyl oligopeptidase inhibitors. Fitoterapia 2015, 103, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Slavík, J.; Slavíková, L. Alkaloids from Corydalis cava (L.) SCHW. et KOERTE. Collect. Czech. Chem. Commun. 1979, 44, 2261–2274. [Google Scholar] [CrossRef]
- Guo, Z.; Cai, R.; Su, H.; Li, Y. Alkaloids in processed rhizoma Corydalis and crude rhizoma Corydalis analyzed by GC/MS. J. Anal. Methods Chem. 2014, 2014, 281342. [Google Scholar] [CrossRef] [PubMed]
- guang Ma, W.; Fukushi, Y.; Tahara, S. Fungitoxic alkaloids from Hokkaido Corydalis species. Fitoterapia 1999, 70, 258–265. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Hošťálková, A.; Kuneš, J.; Macáková, K.; Hrabinová, M.; Opletal, L. Alkaloids from Hydrastidis canadensis and their cholinesterase and prolyl oligopeptidase inhibitory. Ceska Slov. Farm. 2015, 64, 41–43. [Google Scholar] [PubMed]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef]
- Chlebek, J.; Doskocil, I.; Hulcová, D.; Breiterová, K.; Šafratová, M.; Havelek, R.; Habartová, K.; Hošt’álková, A.; Volštátová, T.; Cahlíková, L. Cytotoxicity of naturally occurring isoquinoline alkaloids of different structural types. Nat. Prod. Commun. 2016, 11, 753–756. [Google Scholar] [CrossRef]
- Zhu, J.P. Chinese Materia Medica: Chemistry, Pharmacology and Applications, 1st ed.; CRC Press: Boca Raton, FL, USA, 1998. [Google Scholar]
- Subaiea, G.M.; Aljofan, M.; Devadasu, V.R.; Alshammari, T.M. Acute toxicity testing of newly discovered potential antihepatitis B virus agents of plant origin. Asian J. Pharm. Clin. Res. 2017, 10, 210–213. [Google Scholar]
- Lineweaver, H.; Burk, D. The determination of enzyme dissociation constants. J. Am. Chem. Soc. 1934, 56, 658–666. [Google Scholar] [CrossRef]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef] [PubMed]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crivori, P.; Cruciani, G.; Carrupt, P.A.; Testa, B. Predicting blood-brain barrier permeation from three-dimensional molecular structure. J. Med. Chem. 2000, 43, 2204–2216. [Google Scholar] [CrossRef]
- Cahlíková, L.; Pérez, D.I.; Štěpánková, Š.; Chlebek, J.; Šafratová, M.; Hošt’álková, A.; Opletal, L. In vitro inhibitory effects of 8-O-demethylmaritidine and undulatine on acetylcholinesterase and their predicted penetration across the blood–brain barrier. J. Nat. Prod. 2015, 78, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Muehlbacher, M.; Spitzer, G.M.; Liedl, K.R.; Kornhuber, J. Qualitative prediction of blood–brain barrier permeability on a large and refined dataset. J. Comput. Aided Mol. Des. 2011, 25, 1095–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.H.; Takács-Novák, K.; Mitchell, R.C. On the partition of ampholytes: Application to blood–brain distribution. J. Pharm. Sci. 1997, 86, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, T.S.; Kirshner, D.A.; Lau, E.Y.; Wong, S.E.; Nilmeier, J.P.; Lightstone, F.C. A method to predict blood-brain barrier permeability of drug-like compounds using molecular dynamics simulations. Biophys. J. 2014, 107, 630–641. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Liu, B.; Wang, L.; Jin, Y.-H. An effective PSO-based memetic algorithm for flow shop scheduling. IEEE Trans. Syst. Man. Cybern. B Cybern. 2007, 37, 18–27. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1 and 2 are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 ± SEM (µM) 1 | PAMPA-BBB Permeability (Pe; 10−6 cm·s−1) 3 | LogBB 4 | ||
|---|---|---|---|---|---|
| hAChE | hBChE | SI 2 | |||
| 1 | 0.38 ± 0.05 | >100 | >263 | 2.5 ± 0.1 (CNS±) | −0.100 |
| 2 | 0.70 ± 0.07 | >100 | >143 | 5.0 ± 0.3 (CNS+) | 0.018 |
| Gal 5 | 0.26 ± 0.01 | 18.0 ± 1.90 | 69 | n.d. 6 | n.d. 6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chlebek, J.; Korábečný, J.; Doležal, R.; Štěpánková, Š.; Pérez, D.I.; Hošťálková, A.; Opletal, L.; Cahlíková, L.; Macáková, K.; Kučera, T.; et al. In Vitro and In Silico Acetylcholinesterase Inhibitory Activity of Thalictricavine and Canadine and Their Predicted Penetration across the Blood-Brain Barrier. Molecules 2019, 24, 1340. https://doi.org/10.3390/molecules24071340
Chlebek J, Korábečný J, Doležal R, Štěpánková Š, Pérez DI, Hošťálková A, Opletal L, Cahlíková L, Macáková K, Kučera T, et al. In Vitro and In Silico Acetylcholinesterase Inhibitory Activity of Thalictricavine and Canadine and Their Predicted Penetration across the Blood-Brain Barrier. Molecules. 2019; 24(7):1340. https://doi.org/10.3390/molecules24071340
Chicago/Turabian StyleChlebek, Jakub, Jan Korábečný, Rafael Doležal, Šárka Štěpánková, Daniel I. Pérez, Anna Hošťálková, Lubomír Opletal, Lucie Cahlíková, Kateřina Macáková, Tomáš Kučera, and et al. 2019. "In Vitro and In Silico Acetylcholinesterase Inhibitory Activity of Thalictricavine and Canadine and Their Predicted Penetration across the Blood-Brain Barrier" Molecules 24, no. 7: 1340. https://doi.org/10.3390/molecules24071340