
Synthesis of Pyridine-Dicarboxamide-Cyclohexanone Derivatives: Anticancer and α-Glucosidase Inhibitory Activities and In Silico Study

 ,
,  ,
,  , ,
, ,  and
and
Abstract
:
1. Introduction
2. Results
2.1. Synthesis of 3a–m
2.2. Biological Activities
2.2.1. Anticancer Activity
2.2.2. α-Glucosidase Inhibitory Activity
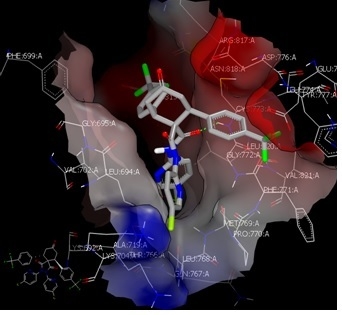
2.3. Molecular Docking Study
2.4. Structure–Activity Relationship (SAR)
3. Materials and Methods
3.1. Experimental
3.2. Anticancer Activity
3.2.1. Cell Lines and Drugs
3.2.2. Cytotoxicity Assay
3.2.3. α-Glucosidase Inhibitory Assay
3.2.4. Molecular Docking Study
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Traoré, T.; Cavagnino, A.; Saettel, N.; Radvanyi, F.; Piguel, S.; Bernard-Pierrot, I.; Stoven, V.; Legraverend, M. New aminopyrimidine derivatives as inhibitors of the TAM family. Eur. J. Med. Chem. 2013, 70, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Alessi, D.R. Kinase drug discovery–what’s next in the field? ACS Chem. Biol. 2012, 8, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Tohyama, O.; Yamaguchi, A.; Matsushima, T.; Takahashi, K.; Funasaka, S.; Shirotori, S.; Asada, M.; Obaishi, H. E7050: A dual c-Met and VEGFR-2 tyrosine kinase inhibitor promotes tumor regression and prolongs survival in mouse xenograft models. Cancer Sci. 2010, 101, 210–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squibb, B.-M. Multiple Ascending Dose Study of BMS-777607 in Subjects With Advanced or Metastatic Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT00605618 (accessed on 15 October 2018).
- Dai, Y.; Siemann, D.W. BMS-777607, a Small-Molecule Met Kinase Inhibitor, Suppresses Hepatocyte Growth Factor–Stimulated Prostate Cancer Metastatic Phenotype In vitro. Mol. Cancer Ther. 2010, 1535–7163. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, G.M.; An, Y.; Cai, Z.-W.; Chen, X.-T.; Clark, C.; Cornelius, L.A.; Dai, J.; Gullo-Brown, J.; Gupta, A.; Henley, B. Discovery of N-(4-(2-amino-3-chloropyridin-4-yloxy)-3-fluorophenyl)-4-ethoxy-1-(4-fluorophenyl)-2-oxo-1, 2-dihydropyridine-3-carboxamide (BMS-777607), a selective and orally efficacious inhibitor of the Met kinase superfamily. J. Med. Chem. 2009, 52, 1251–1254. [Google Scholar] [CrossRef]
- Sharma, S.; Zeng, J.-Y.; Zhuang, C.-M.; Zhou, Y.-Q.; Yao, H.-P.; Hu, X.; Zhang, R.; Wang, M.-H. Small-molecule inhibitor BMS-777607 induces breast cancer cell polyploidy with increased resistance to cytotoxic chemotherapy agents. Mol. Cancer Ther. 2013, 12, 725–736. [Google Scholar] [CrossRef]
- Chu, G.-H.; Gu, M.; Cassel, J.A.; Belanger, S.; Graczyk, T.M.; DeHaven, R.N.; Conway-James, N.; Koblish, M.; Little, P.J.; DeHaven-Hudkins, D.L. Novel malonamide derivatives as potent κ opioid receptor agonists. Bioorg. Med. Chem. Lett. 2007, 17, 1951–1955. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Barakat, A.; Al-Majid, A.M.; Ghabbour, H.A.; Rahman, A.M.; Javaid, K.; Imad, R.; Yousuf, S.; Choudhary, M.I. A concise synthesis and evaluation of new malonamide derivatives as potential α-glucosidase inhibitors. Biorg. Med. Chem. 2016, 24, 1675–1682. [Google Scholar] [CrossRef]
- Barakat, A.; Islam, M.S.; Al-Majid, A.M.; Soliman, S.M.; Ghabbour, H.A.; Yousuf, S.; Choudhary, M.I.; Ul-Haq, Z. Synthesis, molecular structure, spectral analysis and biological activity of new malonamide derivatives as α-glucosidase inhibitors. J. Mol. Struct. 2017, 1134, 253–264. [Google Scholar] [CrossRef]
- Vranken, J.; Dijkgraaf, M.; Kruis, M.; Van Dasselaar, N.; Van der Vegt, M. Iontophoretic administration of S (+)-ketamine in patients with intractable central pain: A placebo-controlled trial. Pain 2005, 118, 224–231. [Google Scholar] [CrossRef]
- Wood, D.M.; Davies, S.; Puchnarewicz, M.; Johnston, A.; Dargan, P.I. Acute toxicity associated with the recreational use of the ketamine derivative methoxetamine. Eur. J. Clin. Pharmacol. 2012, 68, 853–856. [Google Scholar] [CrossRef] [PubMed]
- Gein, V.; Levandovskaya, E.; Nosova, N.; Antonova, N.; Voronina, E.; Vakhrin, M.; Krivenko, A. Synthesis and antibacterial activity of N,N′-diaryl-2-aryl-6-hydroxy-6-methyl-4-oxocyclohexane-1,3-dicarboxamides. Pharm. Chem. J. 2007, 41, 643–645. [Google Scholar] [CrossRef]
- Holland, K.; Naritoku, D.; McKeon, A.; Ferrendelli, J.; Covey, D. Convulsant and anticonvulsant cyclopentanones and cyclohexanones. Mol. Pharmacol. 1990, 37, 98–103. [Google Scholar] [PubMed]
- Liu, L.; Liu, S.; Chen, X.; Guo, L.; Che, Y. Pestalofones A–E, bioactive cyclohexanone derivatives from the plant endophytic fungus Pestalotiopsis fici. Biorg. Med. Chem. 2009, 17, 606–613. [Google Scholar] [CrossRef]
- Adepu, R.; Rambabu, D.; Prasad, B.; Meda, C.L.T.; Kandale, A.; Krishna, G.R.; Reddy, C.M.; Chennuru, L.N.; Parsa, K.V.; Pal, M. Novel thieno [2,3-d] pyrimidines: Their design, synthesis, crystal structure analysis and pharmacological evaluation. Org. Biomol. Chem. 2012, 10, 5554–5569. [Google Scholar] [CrossRef]
- Kumar, T.B.; Dhananjaya, G.; Sumanth, C.; Vaishaly, S.; Botre, G.; Rao, M.S.; Sekhar, K.C.; Kumarc, K.S.; Pal, M. Indion 860 catalyzed cascade reaction: A greener approach to functionalized cyclohexanones and their novel analogues3. RSC Adv. 2013, 3, 2207–2210. [Google Scholar] [CrossRef]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217. [Google Scholar] [CrossRef]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef]
- Aronov, A.M.; Baker, C.; Bemis, G.W.; Cao, J.; Chen, G.; Ford, P.J.; Germann, U.A.; Green, J.; Hale, M.R.; Jacobs, M. Flipped out: Structure-guided design of selective pyrazolylpyrrole ERK inhibitors. J. Med. Chem. 2007, 50, 1280–1287. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal violet assay for determining viability of cultured cells. Cold Spring Harbor Protocols 2016, 2016. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Slater, T.; Sawyer, B.; Sträuli, U. Studies on succinate-tetrazolium reductase systems: III. Points of coupling of four different tetrazolium salts III. Points of coupling of four different tetrazolium salts. Biochim. Biophys. Acta 1963, 77, 383–393. [Google Scholar] [CrossRef]
- Muchmore, S.W.; Souers, A.J.; Akritopoulou-Zanze, I. The use of three-dimensional shape and electrostatic similarity searching in the identification of a melanin-concentrating hormone receptor 1 antagonist. Chem. Biol. Drug Des. 2006, 67, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, K.R.; Fadaly, W.A.; Kamel, G.M.; Elshaier, Y.A.; El-Magd, M.A. Design, synthesis, modeling studies and biological evaluation of thiazolidine derivatives containing pyrazole core as potential anti-diabetic PPAR-γ agonists and anti-inflammatory COX-2 selective inhibitors. Bioorg. Chem. 2019, 82, 86–99. [Google Scholar] [CrossRef]
- Likhitwitayawuid, K.; Angerhofer, C.K.; Chai, H.; Pezzuto, J.M.; Cordell, G.A.; Ruangrungsi, N. Cytotoxic and antimalarial alkaloids from the tubers of Stephania pierrei. J. Nat. Prod. 1993, 56, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Islam, M.S.; Ghawas, H.M.; Al-Majid, A.M.; El-Senduny, F.F.; Badria, F.A.; Elshaier, Y.A.M.; Ghabbour, H.A. Substituted spirooxindole derivatives as potent anticancer agents through inhibition of phosphodiesterase 1. RSC Adv. 2018, 8, 14335–14346. [Google Scholar] [CrossRef]
- Kazeem, M.I.; Ogunbiyi, J.V.; Ashafa, A.O. In vitro studies of the inhibition of a-amylase and α-glucosidase by leaf extracts of Picralimanitida (Stapf). Trop. J. Pharm. Res. 2013, 12, 719–725. [Google Scholar]
- Fast Rigid Exhaustive Docking (FRED) Receptor, version 2.2.5; Open Eye Scientific Software: Santa Fe, NM, USA, 2019; Available online: http://www.eyesopen.com (accessed on 13 March 2019).
- OMEGA, version 2.5.1.4; Open Eye Scientific Software: Santa Fe, NM, USA, 2019; Available online: http://www.eyesopen.com (accessed on 13 March 2019).
- VIDA, version 4.1.2; Open Eye Scientific Software: Santa Fe, NM, USA, 2019; Available online: http://www.eyesopen.com (accessed on 13 March 2019).
Sample Availability: Samples of the compounds 3a–m are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | R | 2a–m | Ar | Product 3a–m | Yield, % |
|---|---|---|---|---|---|
| 1 | H | 2a | C6H5 | 3a | 89 |
| 2 | Cl | 2b | p-CH3C6H4 | 3b | 33 |
| 3 | H | 2c | p-ClC6H4 | 3c | 64 |
| 4 | H | 2d | 2,4-Cl2C6H3 | 3d | 60 |
| 5 | H | 2e | p-BrC6H4 | 3e | 56 |
| 6 | Cl | 2f | m-NO2C6H4 | 3f | 62 |
| 7 | Cl | 2g | p-MeOC6H4 | 3g | 72 |
| 8 | Cl | 2h | β-Naphthalene | 3h | 58 |
| 9 | Cl | 2i | 2-Thiophene | 3i | 60 |
| 10 | Cl | 2j | 2-Furan | 3j | 52 |
| 11 | Cl | 2k | m-BrC6H4 | 3k | 44 |
| 12 | Cl | 2l | p-CF3C6H4 | 3l | 37 |
| 13 | H | 2m | p-FC6H4 | 3m | 85 |
| Compounds a,b | Breast | Oral | Prostate | Colon | Liver | ||
|---|---|---|---|---|---|---|---|
| MCF-7 | MDA-MB-231 | SAS | PC-3 | HCT-116 | HuH-7 | HepG2 | |
| 3a | NA c | NA | NA | NA | NA | NA | NA |
| 3b | NA | NA | NA | NA | NA | NA | NA |
| 3c | 10 ± 0.62 | 7 ± 1.12 | 15 ± 1.3 | 25 ± 1.42 | NA | NA | 8 ± 0.89 |
| 3d | >50 ± 1.17 | 18 ± 0.87 | NA | NA | NA | NA | > 50 |
| 3e | 12 ± 0.54 | 5 ± 0.5 | NA | NA | NA | NA | 8 ± 0.96 |
| 3f | NA | NA | NA | NA | NA | NA | NA |
| 3g | NA | NA | NA | NA | NA | NA | NA |
| 3h | NA | NA | NA | NA | NA | NA | NA |
| 3i | NA | NA | NA | NA | NA | NA | NA |
| 3j | 50 ± 0.78 | 45±3 | NA | NA | NA | NA | >50 ± 1.08 |
| 3k | NA | NA | NA | NA | NA | NA | NA |
| 3l | 18 ± 1.71 | 5 ± 0.25 | 9 ± 0.38 | > 50 ± 2 | 6 ± 0.78 | 4.5 ± 0.3 | 25 ± 0.38 |
| 3m | NA | NA | NA | NA | NA | NA | NA |
| Cisplatin | 9 ± 2.43 | 15 ± 0.71 | 4.5 ± 0.34 | 12 ± 1.25 | 8 ± 0.76 | 14.7 ± 0.5 | 10 ± 0.65 |
| Compounds | IC50 (± SEM μmol/L) |
|---|---|
| 3d | 148.18 ± 3.02 |
| 3i | 418.21 ± 1.02 |
| 3j | 124.24 ± 0.16 |
| Acarbose | 32.71 ± 1.17 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Majid, A.M.; Islam, M.S.; Atef, S.; El-Senduny, F.F.; Badria, F.A.; Elshaier, Y.A.M.M.; Ali, M.; Barakat, A.; Motiur Rahman, A.F.M. Synthesis of Pyridine-Dicarboxamide-Cyclohexanone Derivatives: Anticancer and α-Glucosidase Inhibitory Activities and In Silico Study. Molecules 2019, 24, 1332. https://doi.org/10.3390/molecules24071332
Al-Majid AM, Islam MS, Atef S, El-Senduny FF, Badria FA, Elshaier YAMM, Ali M, Barakat A, Motiur Rahman AFM. Synthesis of Pyridine-Dicarboxamide-Cyclohexanone Derivatives: Anticancer and α-Glucosidase Inhibitory Activities and In Silico Study. Molecules. 2019; 24(7):1332. https://doi.org/10.3390/molecules24071332
Chicago/Turabian StyleAl-Majid, Abdullah Mohammed, Mohammad Shahidul Islam, Saleh Atef, Fardous F. El-Senduny, Farid A. Badria, Yaseen A. M. M. Elshaier, M. Ali, Assem Barakat, and A. F. M. Motiur Rahman. 2019. "Synthesis of Pyridine-Dicarboxamide-Cyclohexanone Derivatives: Anticancer and α-Glucosidase Inhibitory Activities and In Silico Study" Molecules 24, no. 7: 1332. https://doi.org/10.3390/molecules24071332