A Peptide-Based HIV-1 Fusion Inhibitor with Two Tail-Anchors and Palmitic Acid Exhibits Substantially Improved In Vitro and Ex Vivo Anti-HIV-1 Activity and Prolonged In Vivo Half-Life

 , and
, and
Abstract
:1. Introduction
2. Results
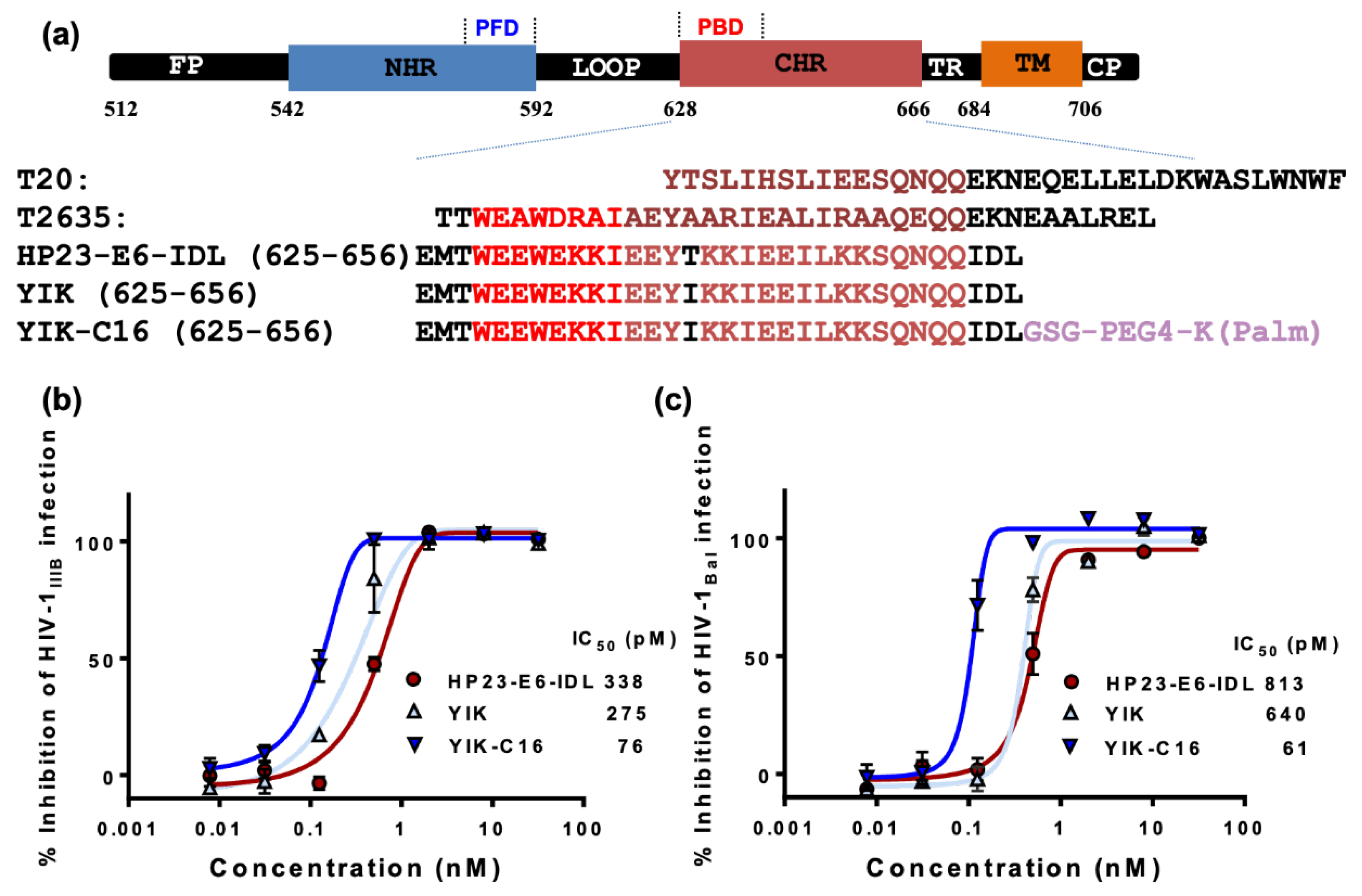
2.1. YIK-C16 was Highly Potent in Inhibiting HIV-1 Infection In Vitro
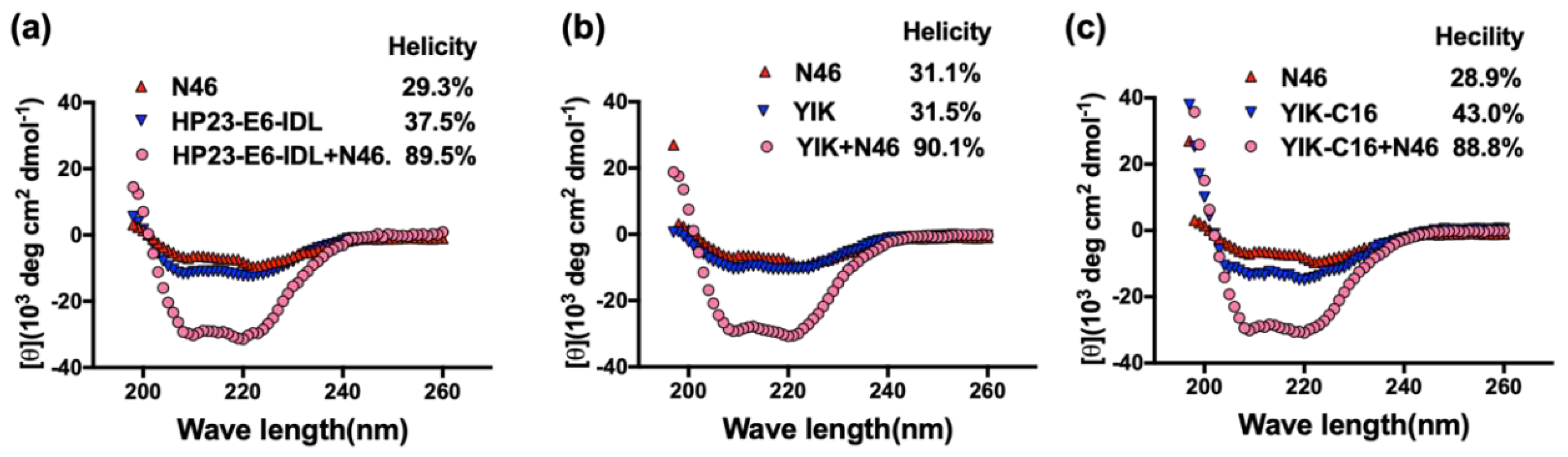
2.2. Secondary Structure of Complex Formed by YIK-C16 and an NHR Peptide
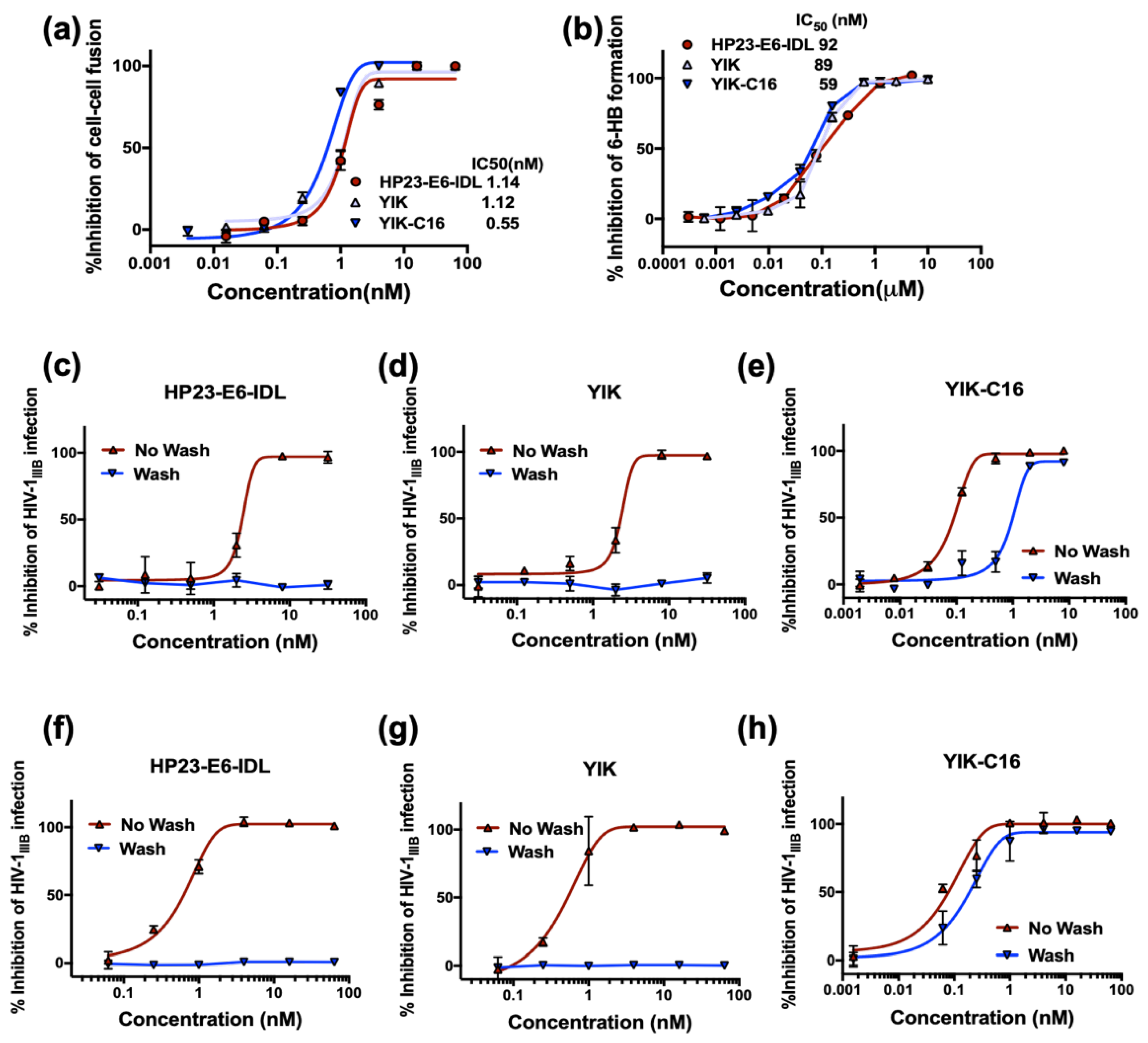
2.3. Improved Anti-HIV-1 Activity of YIK-C16 may Result from its Binding to the Cell Membrane
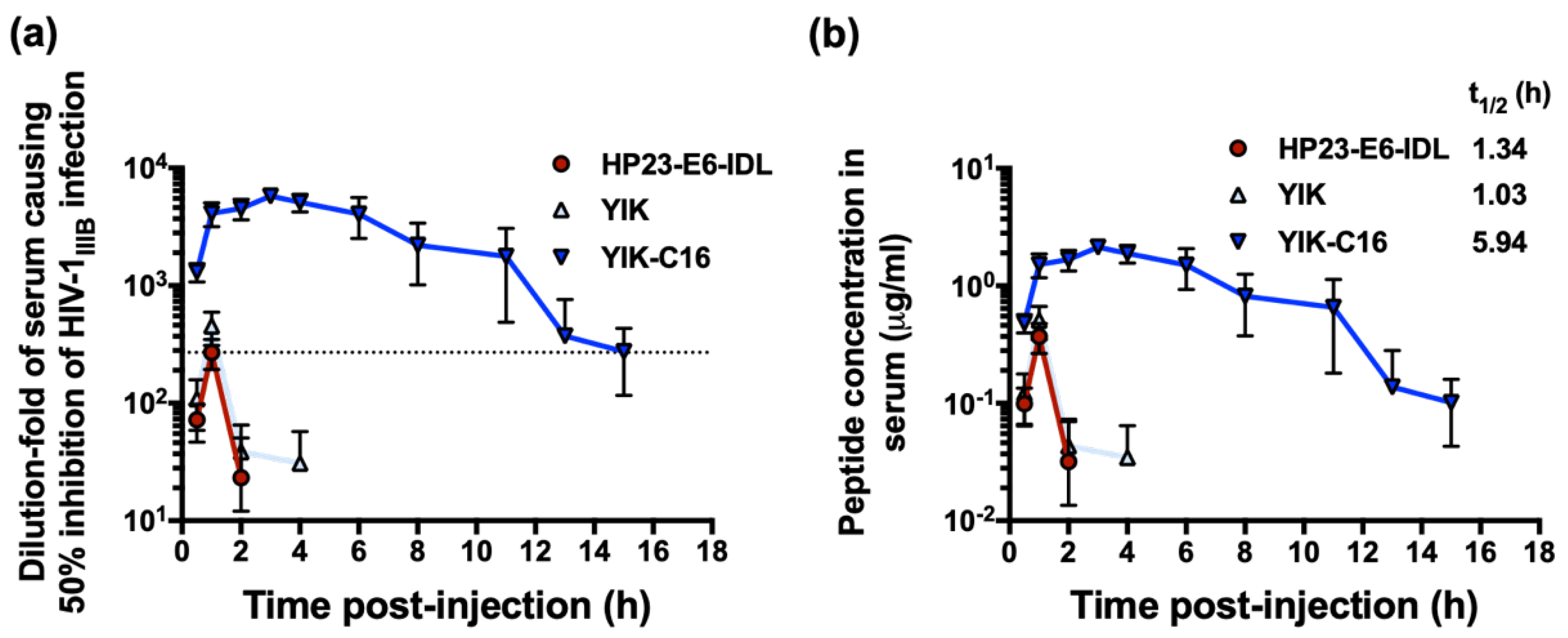
2.4. YIK-C16 Exhibited Improved Ex Vivo Anti-HIV-1 Activity and Prolonged Serum Half-Life
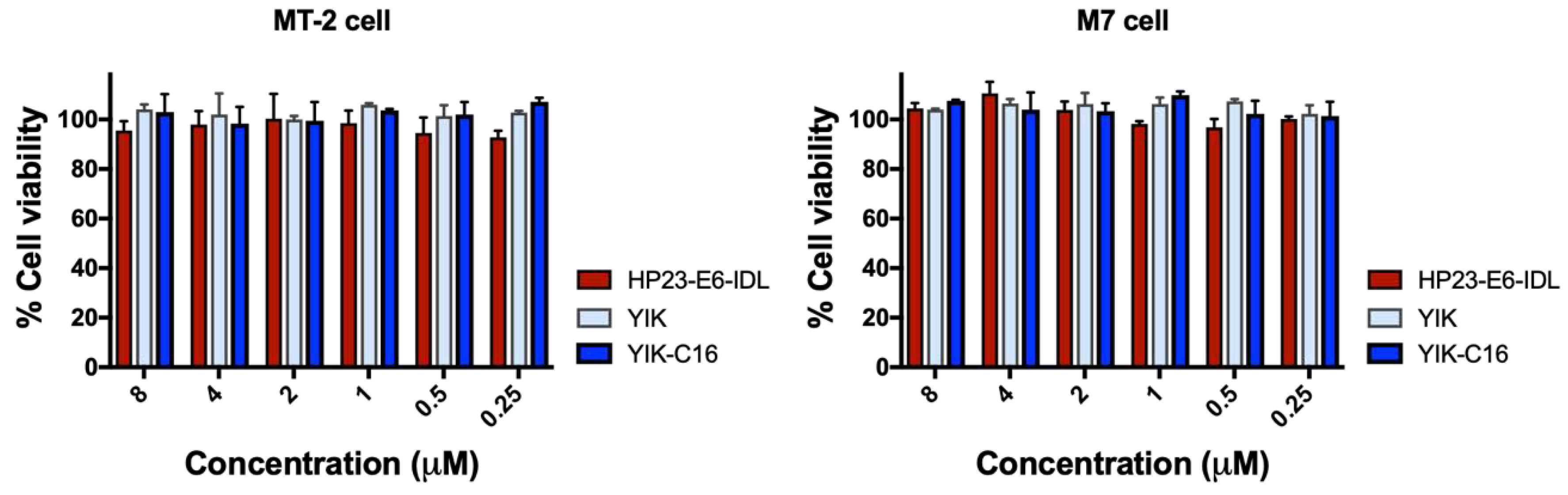
2.5. YIK-C16 Exhibited no In Vitro Cytotoxicity
3. Discussion
4. Materials and Methods
4.1. Peptides, Virus, and Cells
4.2. Inhibition Against HIV-1 Infection by Peptides
4.3. Circular Dichroism (CD) Spectroscopy
4.4. HIV-1-Mediated Cell-Cell Fusion Assay
4.5. Inhibition of 6-HB Formation by Peptides In Vitro
4.6. Ex Vivo Anti-HIV-1 Activity and Serum Half-Life of Peptides
4.7. Cytotoxicity of YIK-C16
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sepkowitz, K.A. Effect of HAART on natural history of AIDS-related opportunistic disorders. Lancet 1998, 351, 228–230. [Google Scholar] [CrossRef]
- Jiang, S.; Lin, K.; Strick, N.; Neurath, A.R. Inhibition of HIV-1 infection by a fusion domain binding peptide from the HIV-1 envelope glycoprotein GP41. Biochem. Biophys. Res. Commun. 1993, 195, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Weissenhorn, W.; Dessen, A.; Harrison, S.C.; Skehel, J.J.; Wiley, D.C. Atomic structure of the ectodomain from HIV-1 gp41. Nature 1997, 387, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Kim, P.S. A trimeric structural subdomain of the HIV-1 transmembrane glycoprotein. J. Biomol. Struct. Dyn. 1997, 15, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Fass, D.; Berger, J.M.; Kim, P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell 1997, 89, 263–273. [Google Scholar] [CrossRef]
- Wild, C.T.; Shugars, D.C.; Greenwell, T.K.; McDanal, C.B.; Matthews, T.J. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA 1994, 91, 9770–9774. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Lin, K.; Strick, N.; Neurath, A.R. HIV-1 inhibition by a peptide. Nature 1993, 365, 113. [Google Scholar] [CrossRef]
- Lalezari, J.P.; Henry, K.; O’Hearn, M.; Montaner, J.S.; Piliero, P.J.; Trottier, B.; Walmsley, S.; Cohen, C.; Kuritzkes, D.R.; Eron, J.J., Jr.; et al. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N. Engl. J. Med. 2003, 348, 2175–2185. [Google Scholar] [CrossRef]
- Patel, I.H.; Zhang, X.; Nieforth, K.; Salgo, M.; Buss, N. Pharmacokinetics, pharmacodynamics and drug interaction potential of enfuvirtide. Clin. Pharmacokinet. 2005, 44, 175–186. [Google Scholar] [CrossRef]
- Su, S.; Zhu, Y.; Ye, S.; Qi, Q.; Xia, S.; Ma, Z.; Yu, F.; Wang, Q.; Zhang, R.; Jiang, S.; et al. Creating an Artificial Tail Anchor as a Novel Strategy To Enhance the Potency of Peptide-Based HIV Fusion Inhibitors. J. Virol. 2017, 91, e01445-16. [Google Scholar] [CrossRef]
- Su, S.; Ma, Z.; Hua, C.; Li, W.; Lu, L.; Jiang, S. Adding an Artificial Tail-Anchor to a Peptide-Based HIV-1 Fusion Inhibitor for Improvement of Its Potency and Resistance Profile. Molecules 2017, 22, 1996. [Google Scholar] [CrossRef] [PubMed]
- Ingallinella, P.; Bianchi, E.; Ladwa, N.A.; Wang, Y.J.; Hrin, R.; Veneziano, M.; Bonelli, F.; Ketas, T.J.; Moore, J.P.; Miller, M.D.; et al. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc. Natl. Acad. Sci. USA 2009, 106, 5801–5806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashkenazi, A.; Viard, M.; Unger, L.; Blumenthal, R.; Shai, Y. Sphingopeptides: Dihydrosphingosine-based fusion inhibitors against wild-type and enfuvirtide-resistant HIV-1. FASEB J. 2012, 26, 4628–4636. [Google Scholar] [CrossRef] [PubMed]
- Augusto, M.T.; Hollmann, A.; Castanho, M.A.; Porotto, M.; Pessi, A.; Santos, N.C. Improvement of HIV fusion inhibitor C34 efficacy by membrane anchoring and enhanced exposure. J. Antimicrob. Chemother. 2014, 69, 1286–1297. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Wang, Q.; Xu, W.; Yu, F.; Hua, C.; Zhu, Y.; Jiang, S.; Lu, L. A novel HIV-1 gp41 tripartite model for rational design of HIV-1 fusion inhibitors with improved antiviral activity. AIDS 2017, 31, 885–894. [Google Scholar] [CrossRef]
- Chong, H.; Zhu, Y.; Yu, D.; He, Y. Structural and Functional Characterization of Membrane Fusion Inhibitors with Extremely Potent Activity against Human Immunodeficiency Virus Type 1 (HIV-1), HIV-2, and Simian Immunodeficiency Virus. J. Virol. 2018, 92, e01088-18. [Google Scholar] [CrossRef]
- Wexler-Cohen, Y.; Shai, Y. Demonstrating the C-terminal boundary of the HIV 1 fusion conformation in a dynamic ongoing fusion process and implication for fusion inhibition. FASEB J. 2007, 21, 3677–3684. [Google Scholar] [CrossRef] [PubMed]
- Wexler-Cohen, Y.; Shai, Y. Membrane-anchored HIV-1 N-heptad repeat peptides are highly potent cell fusion inhibitors via an altered mode of action. PLoS Pathog. 2009, 5, e1000509. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.; Xue, J.; Zhu, Y.; Cong, Z.; Chen, T.; Wei, Q.; Qin, C.; He, Y. Monotherapy with a low-dose lipopeptide HIV fusion inhibitor maintains long-term viral suppression in rhesus macaques. PLoS Pathog. 2019, 15, e1007552. [Google Scholar] [CrossRef]
- Allen, G.D. MODFIT: A pharmacokinetics computer program. Biopharm. Drug Dispos. 1990, 11, 477–498. [Google Scholar] [CrossRef]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Viard, M.; Parolini, I.; Sargiacomo, M.; Fecchi, K.; Ramoni, C.; Ablan, S.; Ruscetti, F.W.; Wang, J.M.; Blumenthal, R. Role of cholesterol in human immunodeficiency virus type 1 envelope protein-mediated fusion with host cells. J. Virol. 2002, 76, 11584–11595. [Google Scholar] [CrossRef] [PubMed]
- Guyader, M.; Kiyokawa, E.; Abrami, L.; Turelli, P.; Trono, D. Role for human immunodeficiency virus type 1 membrane cholesterol in viral internalization. J. Virol. 2002, 76, 10356–10364. [Google Scholar] [CrossRef] [PubMed]
- Ono, A.; Freed, E.O. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. USA 2001, 98, 13925–13930. [Google Scholar] [CrossRef] [Green Version]
- Liao, Z.; Cimakasky, L.M.; Hampton, R.; Nguyen, D.H.; Hildreth, J.E. Lipid rafts and HIV pathogenesis: Host membrane cholesterol is required for infection by HIV type 1. AIDS Res. Hum. Retrovir. 2001, 17, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Graham, D.R.; Hildreth, J.E. Lipid rafts and HIV pathogenesis: Virion-associated cholesterol is required for fusion and infection of susceptible cells. AIDS Res. Hum. Retrovir. 2003, 19, 675–687. [Google Scholar] [CrossRef]
- Rawat, S.S.; Viard, M.; Gallo, S.A.; Blumenthal, R.; Puri, A. Sphingolipids, cholesterol, and HIV-1: A paradigm in viral fusion. Glycoconj. J. 2006, 23, 189–197. [Google Scholar] [CrossRef]
- Liu, S.; Lu, H.; Niu, J.; Xu, Y.; Wu, S.; Jiang, S. Different from the HIV fusion inhibitor C34, the anti-HIV drug Fuzeon (T-20) inhibits HIV-1 entry by targeting multiple sites in gp41 and gp120. J. Biol. Chem. 2005, 280, 11259–11273. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Wexler-Cohen, Y.; Shai, Y. Multifaceted action of Fuzeon as virus-cell membrane fusion inhibitor. Biochim. Biophys. Acta 2011, 1808, 2352–2358. [Google Scholar] [CrossRef]
- Veiga, S.; Henriques, S.; Santos, N.C.; Castanho, M. Putative role of membranes in the HIV fusion inhibitor enfuvirtide mode of action at the molecular level. Biochem. J. 2004, 377, 107–110. [Google Scholar] [CrossRef] [Green Version]
- Wexler-Cohen, Y.; Ashkenazi, A.; Viard, M.; Blumenthal, R.; Shai, Y. Virus-cell and cell-cell fusion mediated by the HIV-1 envelope glycoprotein is inhibited by short gp41 N-terminal membrane-anchored peptides lacking the critical pocket domain. FASEB J. 2010, 24, 4196–4202. [Google Scholar] [CrossRef] [Green Version]
- Oleszko, A.; Hartwich, J.; Gasior-Glogowska, M.; Olsztynska-Janus, S. Changes of albumin secondary structure after palmitic acid binding. FT-IR spectroscopic study. Acta Bioeng. Biomech. 2018, 20, 59–64. [Google Scholar]
- Cho, J.; Lim, S.I.; Yang, B.S.; Hahn, Y.S.; Kwon, I. Generation of therapeutic protein variants with the human serum albumin binding capacity via site-specific fatty acid conjugation. Sci. Rep. 2017, 7, 18041. [Google Scholar] [CrossRef] [PubMed]
- Abu Ajaj, K.; Graeser, R.; Fichtner, I.; Kratz, F. In vitro and in vivo study of an albumin-binding prodrug of doxorubicin that is cleaved by cathepsin B. Cancer Chemother. Pharmacol. 2009, 64, 413–418. [Google Scholar] [CrossRef]
- Xie, D.; Yao, C.; Wang, L.; Min, W.; Xu, J.; Xiao, J.; Huang, M.; Chen, B.; Liu, B.; Li, X.; et al. An albumin-conjugated peptide exhibits potent anti-HIV activity and long in vivo half-life. Antimicrob. Agents Chemother. 2010, 54, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.; Yao, X.; Zhang, C.; Cai, L.; Cui, S.; Wang, Y.; He, Y. Biophysical property and broad anti-HIV activity of albuvirtide, a 3-maleimimidopropionic acid-modified peptide fusion inhibitor. PLoS ONE 2012, 7, e32599. [Google Scholar] [CrossRef] [PubMed]
- Guenaga, J.; Garces, F.; de Val, N.; Stanfield, R.L.; Dubrovskaya, V.; Higgins, B.; Carrette, B.; Ward, A.B.; Wilson, I.A.; Wyatt, R.T. Glycine Substitution at Helix-to-Coil Transitions Facilitates the Structural Determination of a Stabilized Subtype C HIV Envelope Glycoprotein. Immunity 2017, 46, 792–803.e3. [Google Scholar] [CrossRef]
- Lu, L.; Tong, P.; Yu, X.; Pan, C.; Zou, P.; Chen, Y.H.; Jiang, S. HIV-1 variants with a single-point mutation in the gp41 pocket region exhibiting different susceptibility to HIV fusion inhibitors with pocket- or membrane-binding domain. Biochim. Biophys. Acta 2012, 1818, 2950–2957. [Google Scholar] [CrossRef] [Green Version]
- Qi, Q.; Wang, Q.; Chen, W.; Yu, F.; Du, L.; Dimitrov, D.S.; Lu, L.; Jiang, S. Anti-HIV antibody and drug combinations exhibit synergistic activity against drug-resistant HIV-1 strains. J. Infect. 2017, 75, 68–71. [Google Scholar] [CrossRef]
- Eggink, D.; Bontjer, I.; Langedijk, J.P.; Berkhout, B.; Sanders, R.W. Resistance of human immunodeficiency virus type 1 to a third-generation fusion inhibitor requires multiple mutations in gp41 and is accompanied by a dramatic loss of gp41 function. J. Virol. 2011, 85, 10785–10797. [Google Scholar] [CrossRef]
- Su, Y.; Chong, H.; Qiu, Z.; Xiong, S.; He, Y. Mechanism of HIV-1 Resistance to Short-Peptide Fusion Inhibitors Targeting the Gp41 Pocket. J. Virol. 2015, 89, 5801–5811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Bi, W.; Zhu, X.; Li, H.; Qi, Q.; Yu, F.; Lu, L.; Jiang, S. Nonneutralizing Antibodies Induced by the HIV-1 gp41 NHR Domain Gain Neutralizing Activity in the Presence of the HIV Fusion Inhibitor Enfuvirtide: A Potential Therapeutic Vaccine Strategy. J. Virol. 2015, 89, 6960–6964. [Google Scholar] [CrossRef] [PubMed]
- Si, L.; Meng, K.; Tian, Z.; Sun, J.; Li, H.; Zhang, Z.; Soloveva, V.; Li, H.; Fu, G.; Xia, Q.; et al. Triterpenoids manipulate a broad range of virus-host fusion via wrapping the HR2 domain prevalent in viral envelopes. Sci. Adv. 2018, 4, eaau8408. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhu, Y.; Wang, Q.; Ye, L.; Dai, Y.; Su, S.; Yu, F.; Ying, T.; Yang, C.; Jiang, S.; et al. An immunogen containing four tandem 10E8 epitope repeats with exposed key residues induces antibodies that neutralize HIV-1 and activates an ADCC reporter gene. Emerg. Microbes Infect. 2016, 5, e65. [Google Scholar] [CrossRef]
- Zhu, X.; Zhu, Y.; Ye, S.; Wang, Q.; Xu, W.; Su, S.; Sun, Z.; Yu, F.; Liu, Q.; Wang, C.; et al. Improved Pharmacological and Structural Properties of HIV Fusion Inhibitor AP3 over Enfuvirtide: Highlighting Advantages of Artificial Peptide Strategy. Sci. Rep. 2015, 5, 13028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Qian, H.; Miyamoto, F.; Naito, T.; Kawaji, K.; Kajiwara, K.; Hattori, T.; Matsuoka, M.; Watanabe, K.; Oishi, S.; et al. A simple, rapid, and sensitive system for the evaluation of anti-viral drugs in rats. Biochem. Biophys. Res. Commun. 2012, 424, 257–261. [Google Scholar] [CrossRef]
- Yang, W.; Sun, Z.; Hua, C.; Wang, Q.; Xu, W.; Deng, Q.; Pan, Y.; Lu, L.; Jiang, S. Chidamide, a histone deacetylase inhibitor-based anticancer drug, effectively reactivates latent HIV-1 provirus. Microbes Infect. 2018, 20, 626–634. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viruses | a IC50 (pM) | ||
|---|---|---|---|
| HP23-E6-IDL | YIK | YIK-C16 | |
| T20-Resistant Strains | |||
| HIV-1 NL4-3 D36G (WT) | 912 ± 29 | 784 ± 32 | 69 ± 8 |
| (D36G) V38A | 647 ± 24 | 627 ± 25 | 99 ± 4 |
| (D36G) V38A, N42D | 1543 ± 253 | 1423 ± 184 | 179 ± 4 |
| (D36G) V38E, N42S | 748 ± 12 | 708 ± 50 | 68 ± 6 |
| (D36G) V38A, N42T | 838 ± 31 | 771 ± 21 | 40 ± 3 |
| (D36G) N42T, N43K | 941 ± 19 | 769 ± 22 | 60 ± 7 |
| T2635-Resistant Strains | |||
| HIV-1 LAI (WT) | 932 ± 59 | 460 ± 18 | 83 ± 3 |
| A6V | 349 ± 19 | 773 ± 26 | 106 ± 7 |
| Q66R | 496 ± 168 | 707 ± 216 | 65 ± 10 |
| K90E | 1423 ± 242 | 885 ± 288 | 98 ± 6 |
| K154Q | 1246 ± 249 | 1490 ± 401 | 71 ± 7 |
| Q79E/N126K | 1312 ± 59 | 1150 ± 263 | 84 ± 8 |
| K90E/N126K | 842 ± 66 | 797 ± 22 | 81 ± 3 |
| HP23-Resistant Strains | |||
| HIV-1 NL4-3 (WT) | 699 ± 25 | 856 ± 30 | 65 ± 8 |
| E49K | 2045 ± 126 | 1823 ± 72 | 115 ± 21 |
| E49K/N126K | 2563 ± 318 | 1378 ± 228 | 81 ± 13 |
| D36G/E49K/N126K | 3142 ± 400 | 2823 ± 611 | 100 ± 11 |
| L34S/D36G/E49K/E136G | 4937 ± 1100 | 5286 ± 569 | 188 ± 33 |
| Parameter | Unit | Mice (i.p., n = 3) |
|---|---|---|
| Tmax | h | 4.0 ± 1.7 |
| Cmax | μg/mL | 2.2 ± 0.1 |
| t½ | h | 5.9 ± 3.2 |
| AUC0-15h | h*μg/mL | 14.9 ± 3.8 |
| AUCINF_obs | h*μg/mL | 15.0 ± 3.7 |
| Vz_F_obs/Vz_obs | mL/kg | 2738.8 ± 740.7 |
| Cl_F_obs/Cl_obs | mL/h/kg | 346.4 ± 76.1 |
| MRTlast | h | 9.5 ± 5.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, S.; Rasquinha, G.; Du, L.; Wang, Q.; Xu, W.; Li, W.; Lu, L.; Jiang, S. A Peptide-Based HIV-1 Fusion Inhibitor with Two Tail-Anchors and Palmitic Acid Exhibits Substantially Improved In Vitro and Ex Vivo Anti-HIV-1 Activity and Prolonged In Vivo Half-Life. Molecules 2019, 24, 1134. https://doi.org/10.3390/molecules24061134
Su S, Rasquinha G, Du L, Wang Q, Xu W, Li W, Lu L, Jiang S. A Peptide-Based HIV-1 Fusion Inhibitor with Two Tail-Anchors and Palmitic Acid Exhibits Substantially Improved In Vitro and Ex Vivo Anti-HIV-1 Activity and Prolonged In Vivo Half-Life. Molecules. 2019; 24(6):1134. https://doi.org/10.3390/molecules24061134
Chicago/Turabian StyleSu, Shan, Giselle Rasquinha, Lanying Du, Qian Wang, Wei Xu, Weihua Li, Lu Lu, and Shibo Jiang. 2019. "A Peptide-Based HIV-1 Fusion Inhibitor with Two Tail-Anchors and Palmitic Acid Exhibits Substantially Improved In Vitro and Ex Vivo Anti-HIV-1 Activity and Prolonged In Vivo Half-Life" Molecules 24, no. 6: 1134. https://doi.org/10.3390/molecules24061134