
Computer-Aided Discovery of Small Molecules Targeting the RNA Splicing Activity of hnRNP A1 in Castration-Resistant Prostate Cancer

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
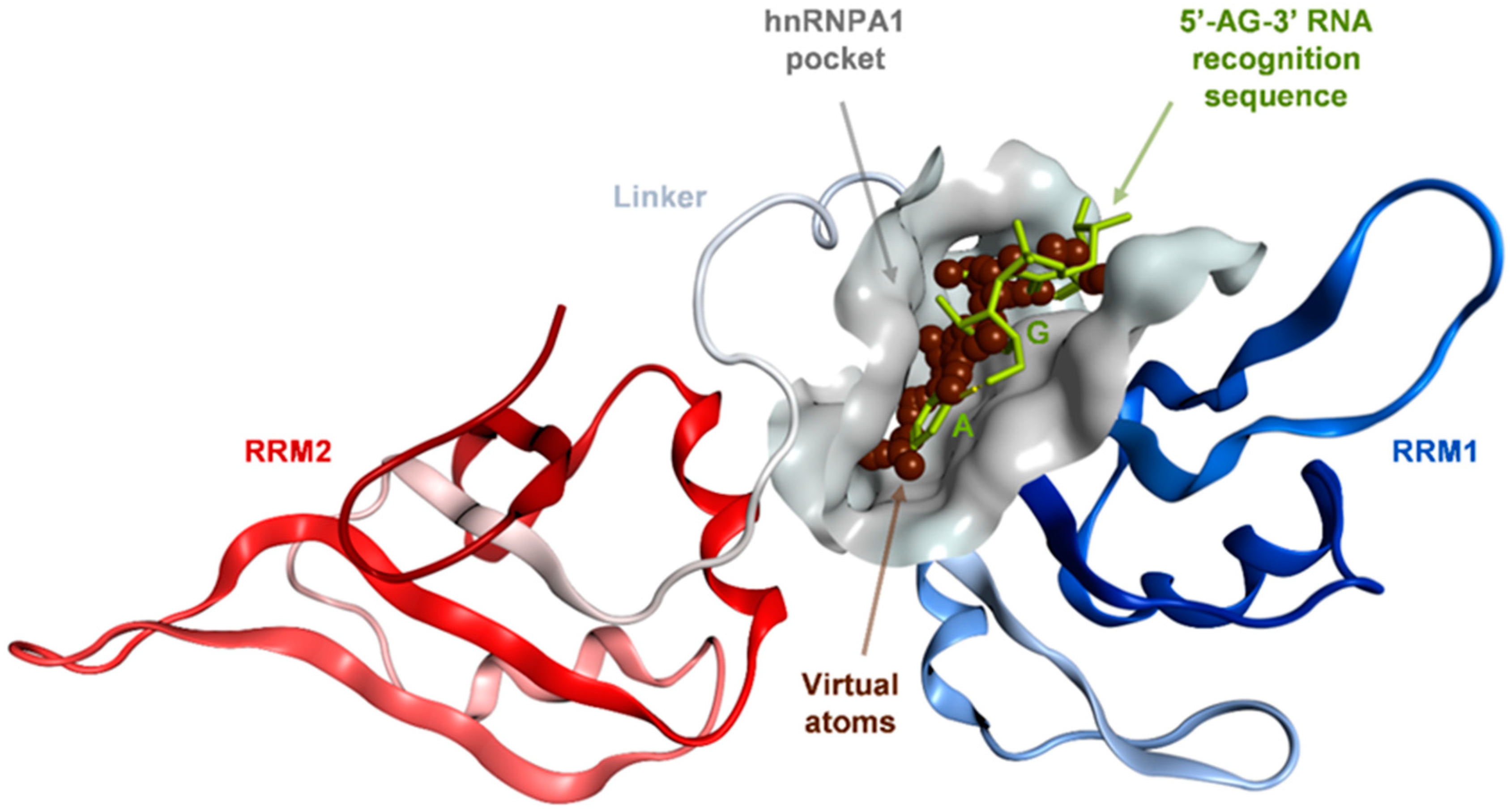
2.1. Binding Site Identification on hnRNP A1 RBD
2.2. In Silico Identification of Hit Compounds Targeting the hnRNP A1 RBD Site
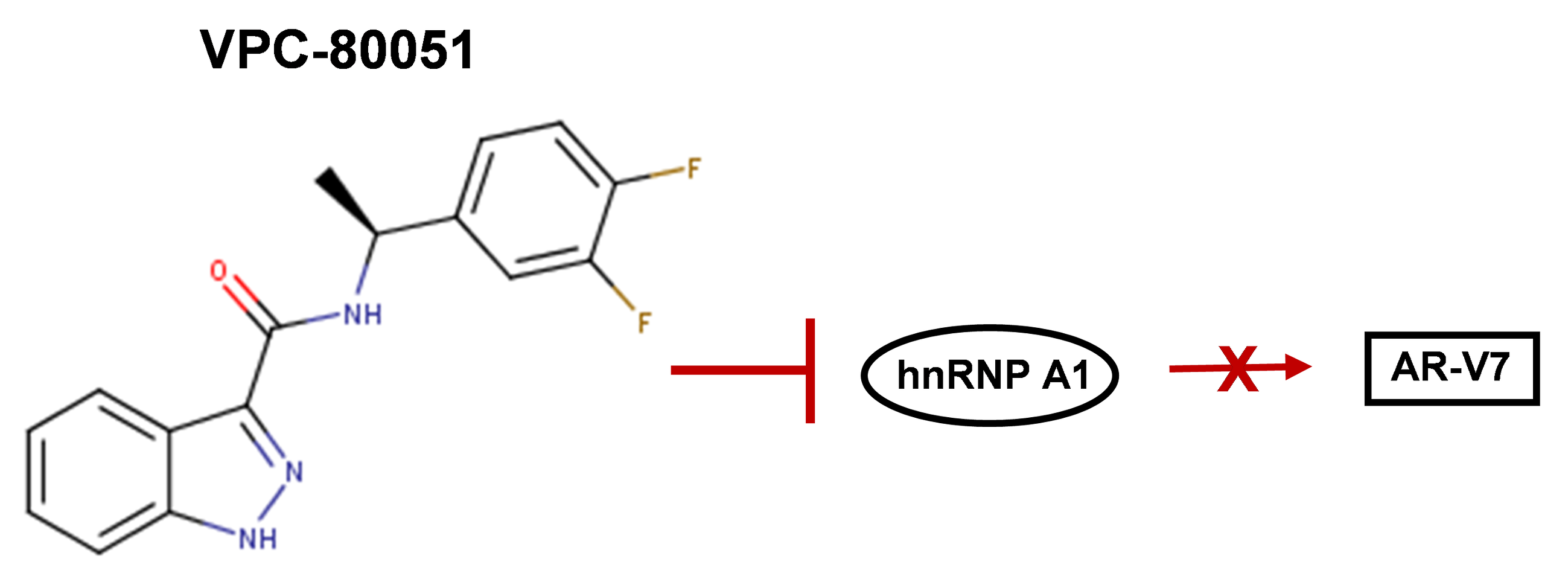
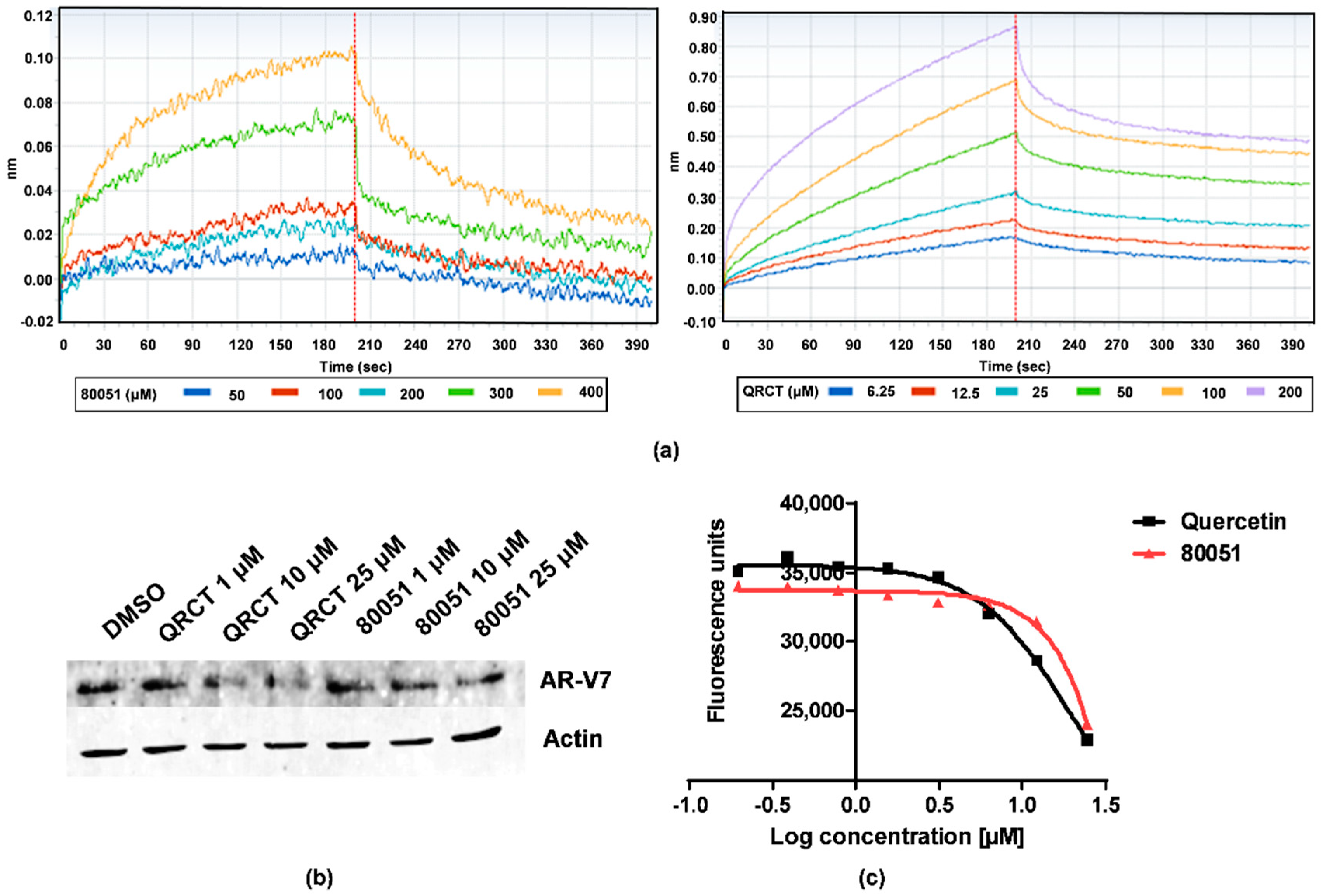
2.3. In Vitro Characterization of Hit Compound VPC-80051
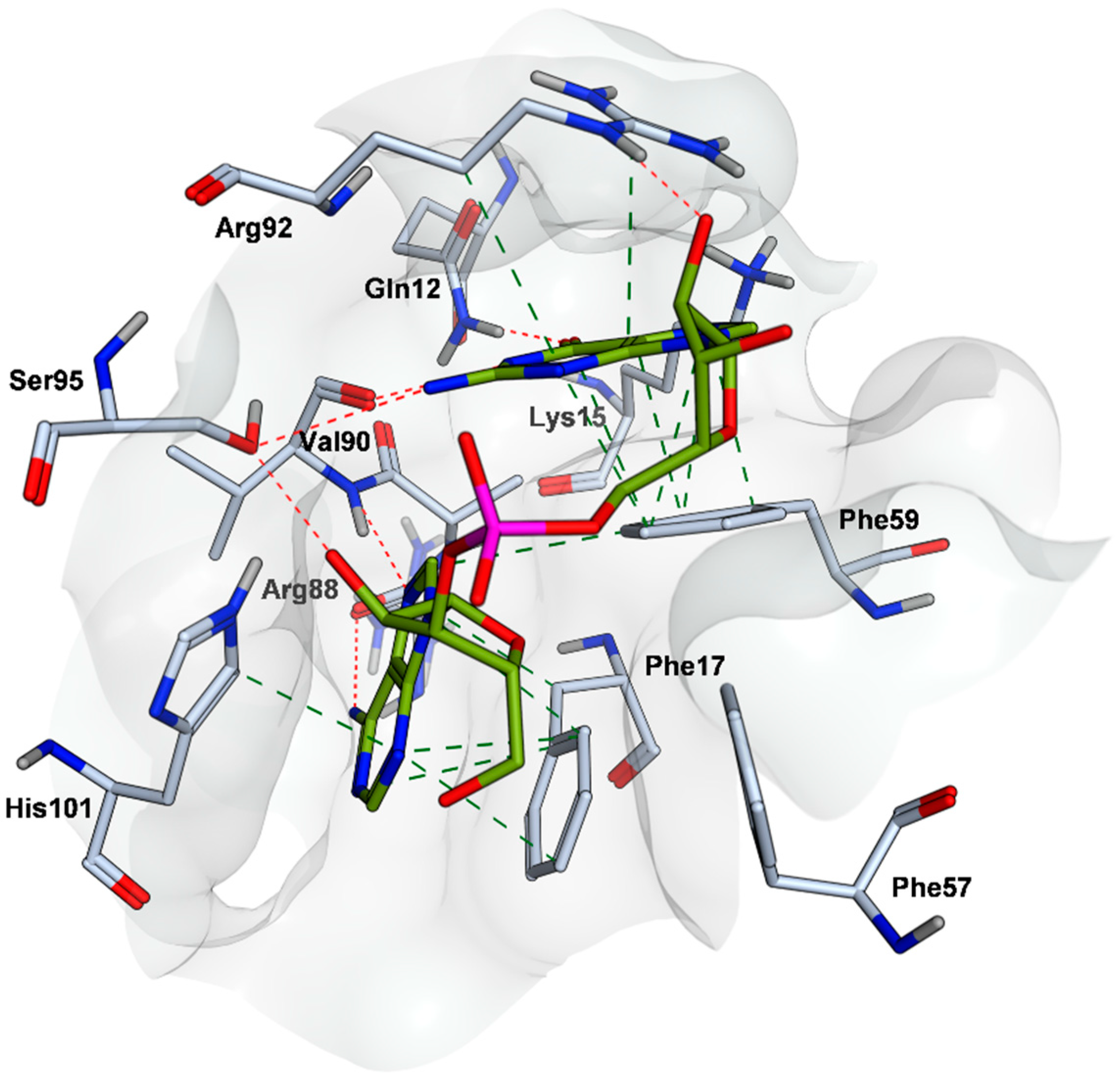
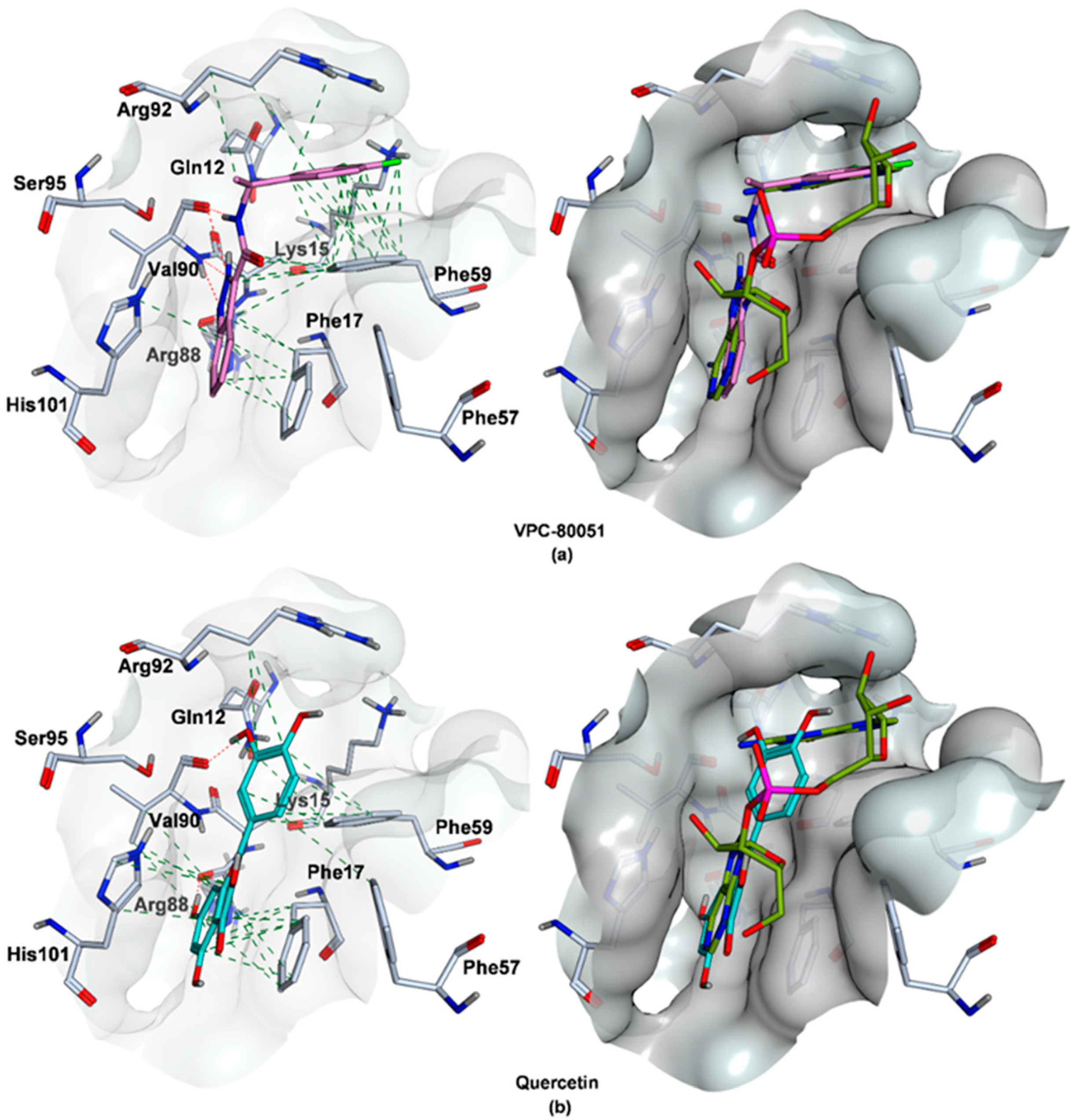
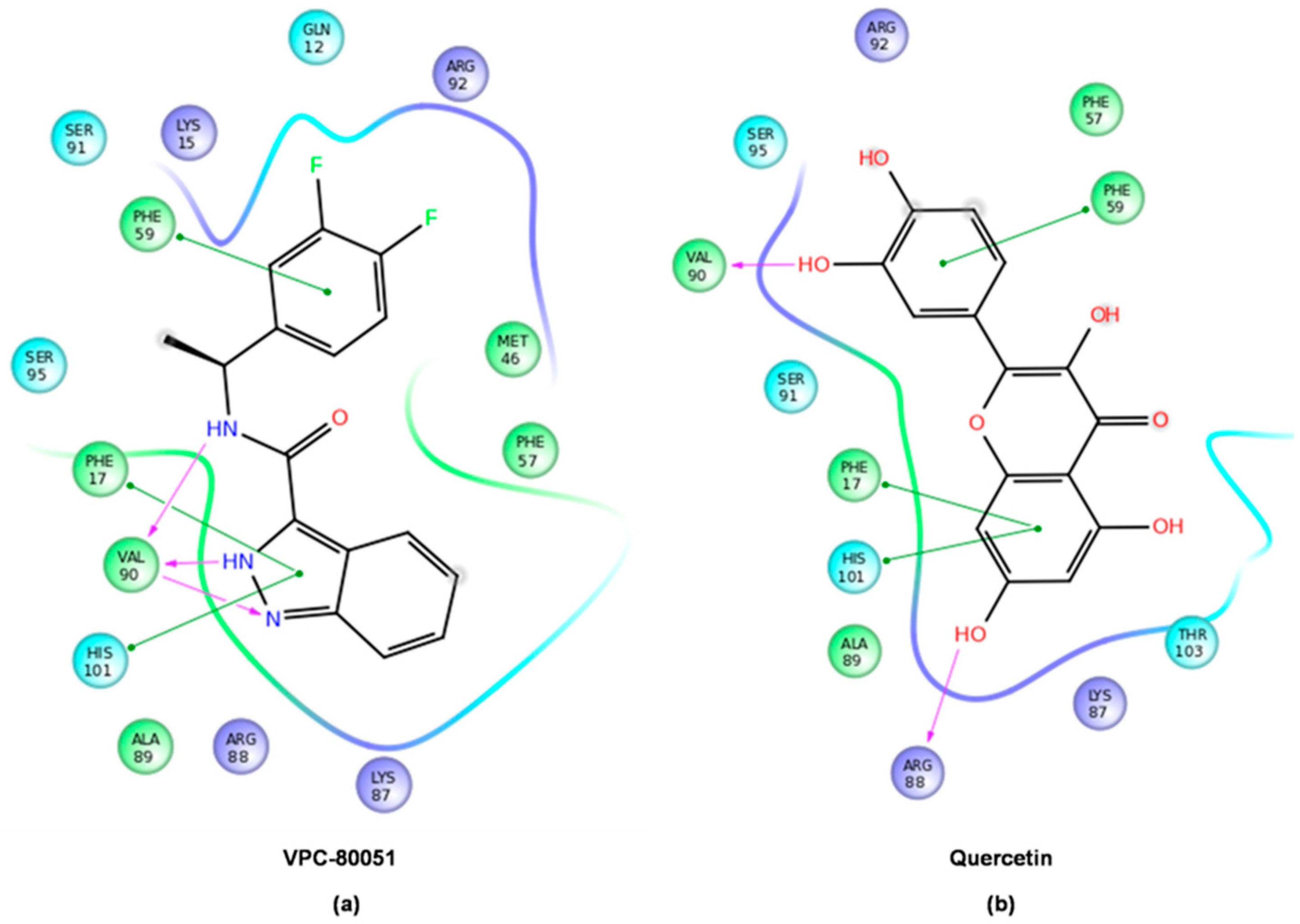
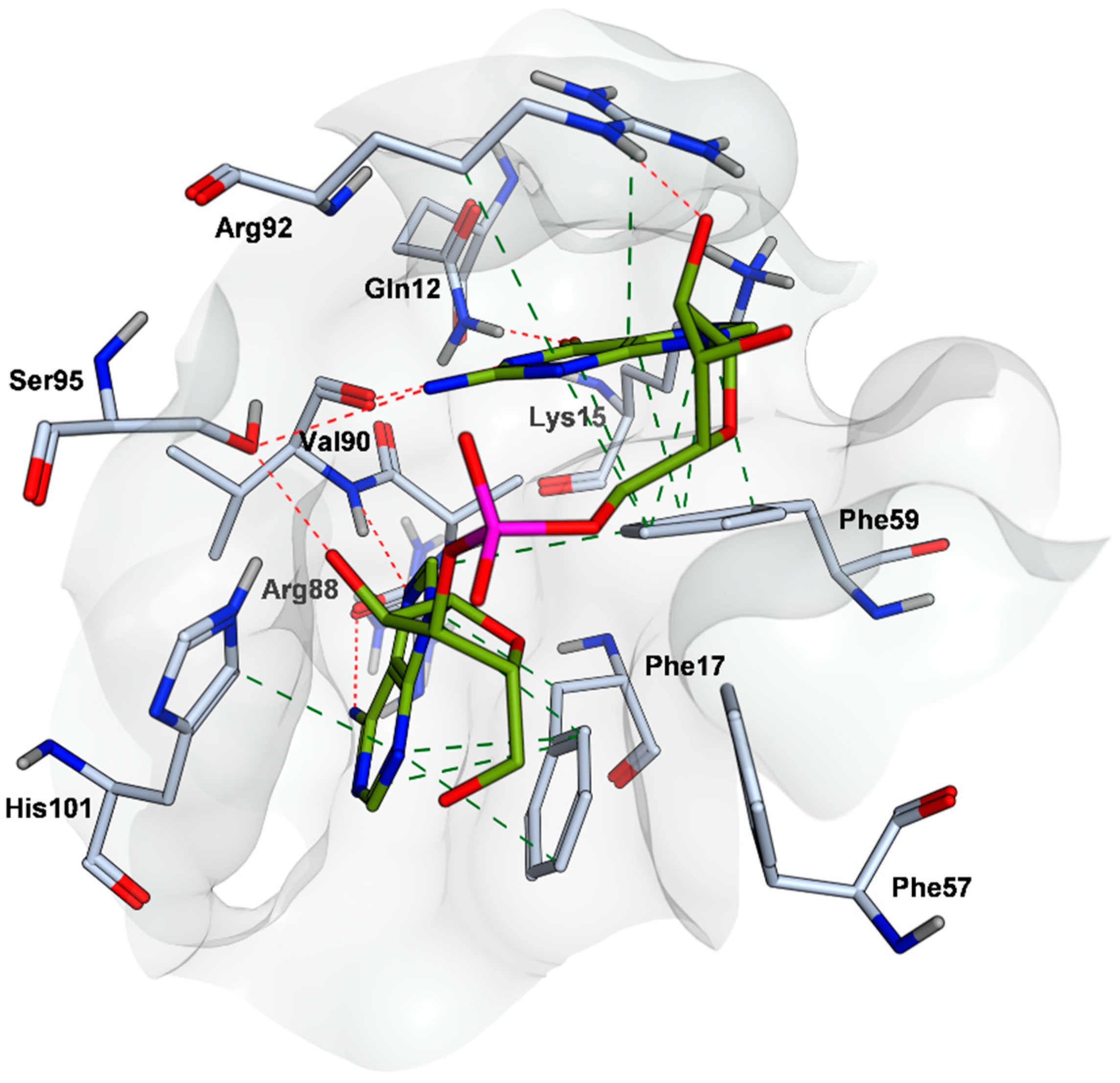
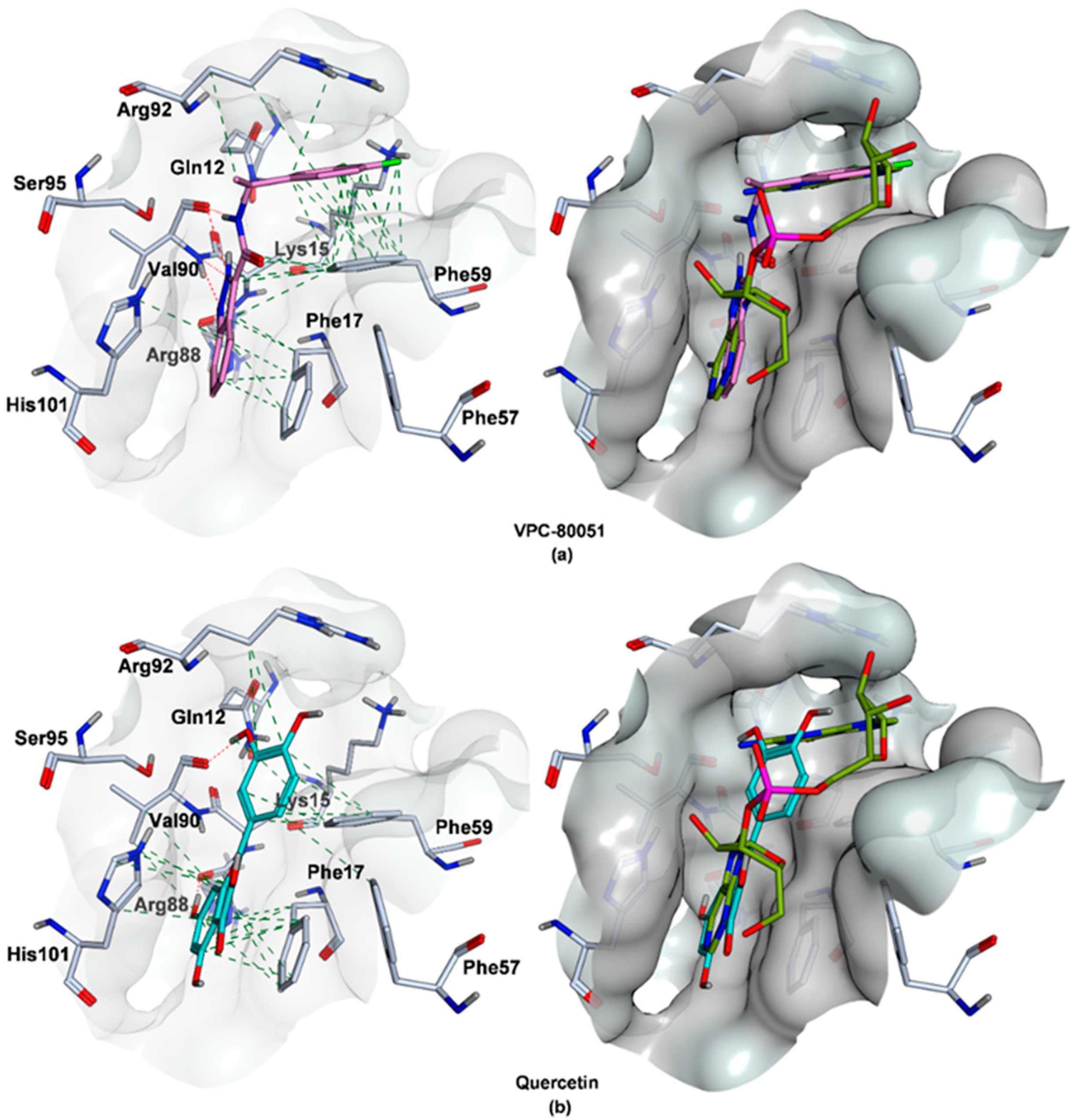
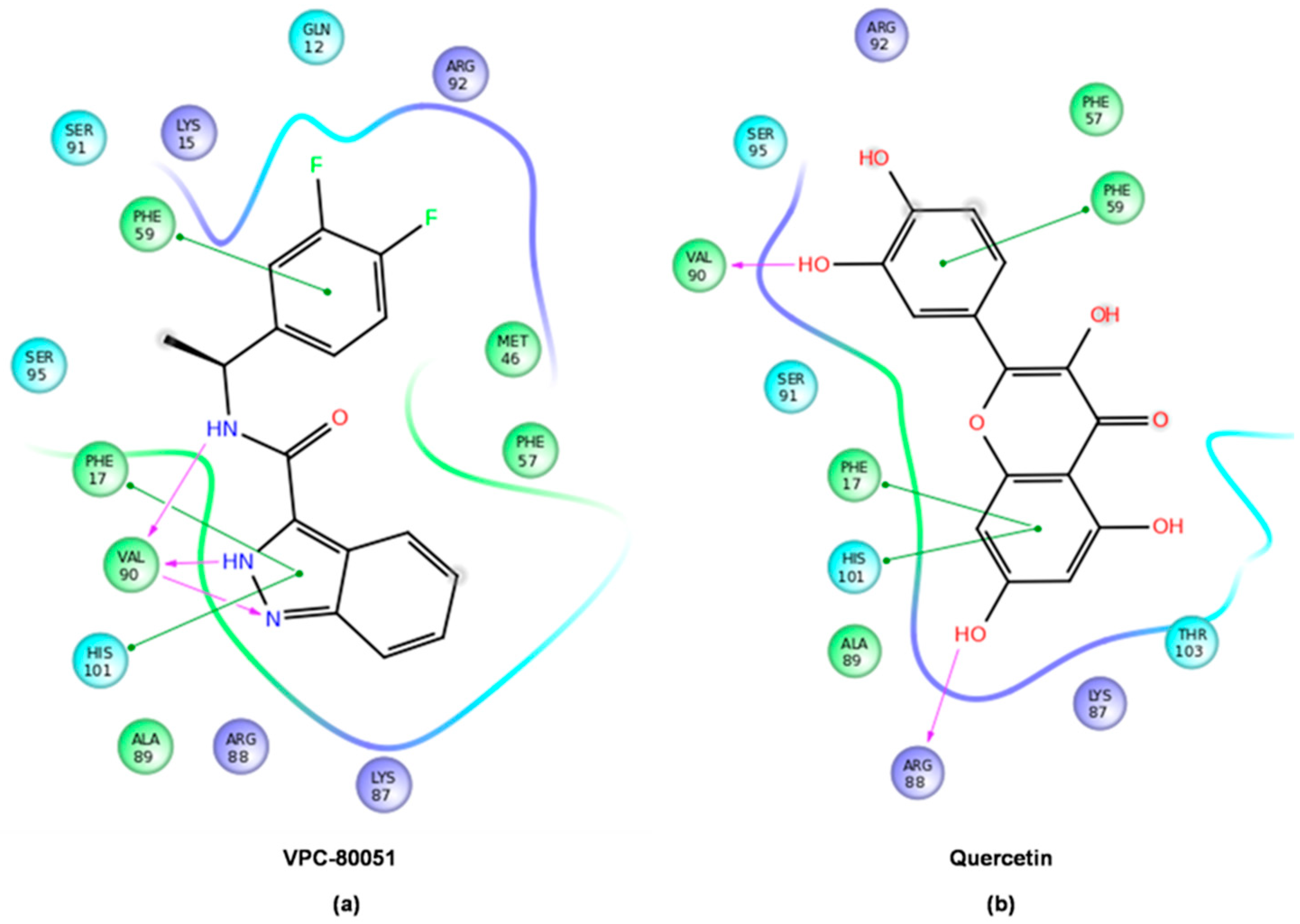
2.4. Mode of Binding of VPC-80051 to hnRNP A1 RBD Site
3. Discussion
4. Materials and Methods
4.1. Binding Site Identification on hnRNP A1 RBD
4.2. Virtual Screening
4.3. MM/GBSA Simulations
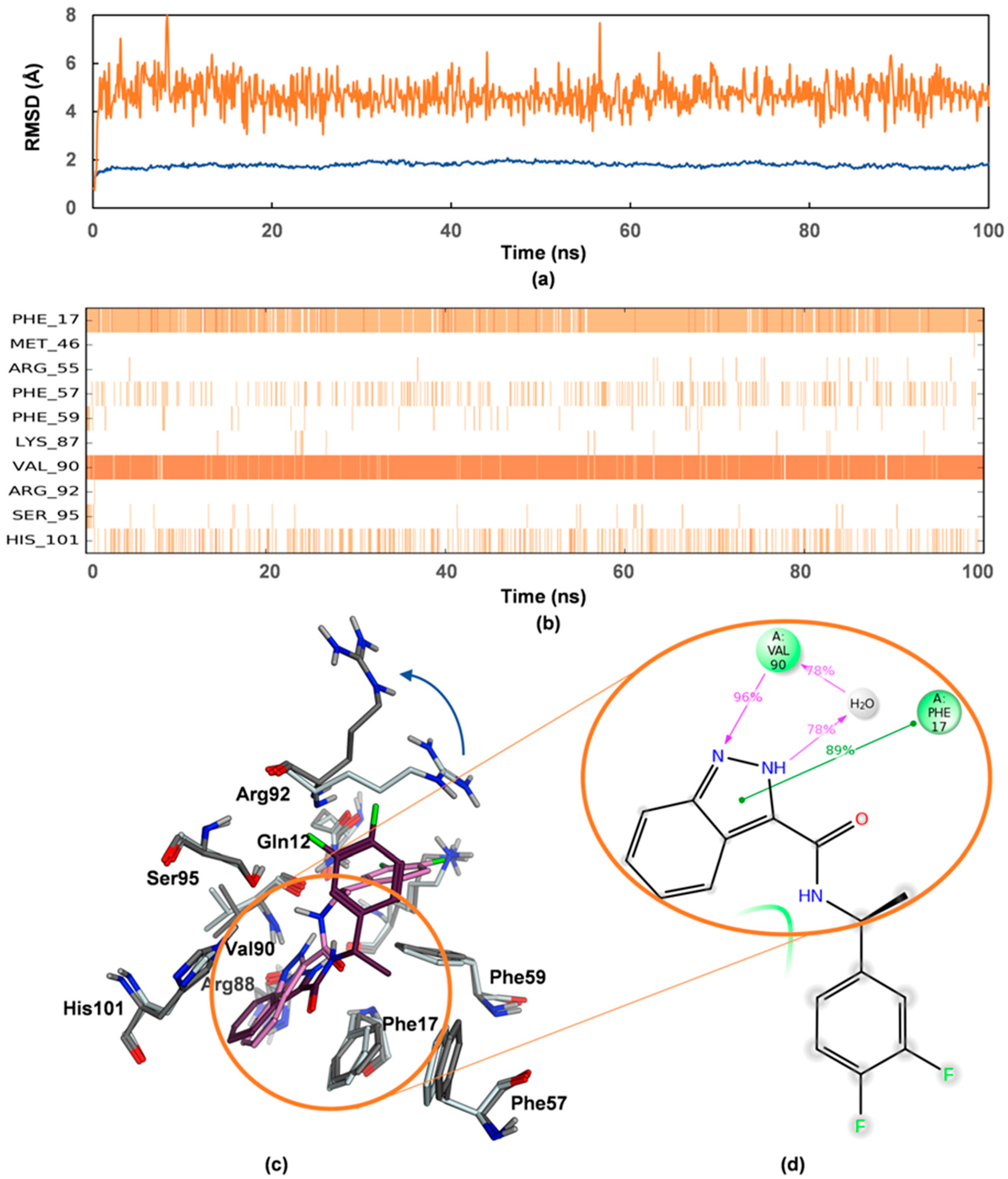
4.4. Molecular Dynamics Simulations
4.5. AR-V7 Level Measurement
4.6. Bio-Layer Interferometry Assay
4.7. qRT-PCR
4.8. Western Blotting
4.9. Cell Viability Assay
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roy, R.; Huang, Y.; Seckl, M.J.; Pardo, O.E. Emerging roles of hnRNPA1 in modulating malignant transformation. Wiley Interdiscip. Rev. RNA 2017, 8, e1431. [Google Scholar] [CrossRef] [PubMed]
- Jean-Philippe, J.; Paz, S.; Caputi, M. hnRNP A1: The Swiss army knife of gene expression. Int. J. Mol. Sci. 2013, 14, 18999–19024. [Google Scholar] [CrossRef] [PubMed]
- Lapuk, A.V.; Volik, S.V.; Wang, Y.; Collins, C.C. The role of mRNA splicing in prostate cancer. Asian J. Androl. 2014, 16, 515–521. [Google Scholar] [CrossRef]
- Nadiminty, N.; Tummala, R.; Liu, C.; Lou, W.; Evans, C.P.; Gao, A.C. NF-kappaB2/p52:c-Myc:hnRNPA1 Pathway Regulates Expression of Androgen Receptor Splice Variants and Enzalutamide Sensitivity in Prostate Cancer. Mol. Cancer Ther. 2015, 14, 1884–1895. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhou, Y.; Lou, Y.; Zhong, H. Knockdown of HNRNPA1 inhibits lung adenocarcinoma cell proliferation through cell cycle arrest at G0/G1 phase. Gene 2016, 576, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, J.; Wang, W.; Xiang, L.; Wang, J.; Liu, S.; Zhou, H.; Guo, Z. High expression of hnRNPA1 promotes cell invasion by inducing EMT in gastric cancer. Oncol. Rep. 2018, 39, 1693–1701. [Google Scholar] [CrossRef] [PubMed]
- Loh, T.J.; Moon, H.; Cho, S.; Jang, H.; Liu, Y.C.; Tai, H.; Jung, D.W.; Williams, D.R.; Kim, H.R.; Shin, M.G.; et al. CD44 alternative splicing and hnRNP A1 expression are associated with the metastasis of breast cancer. Oncol. Rep. 2015, 34, 1231–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockstedt, E.; Rickers, A.; Kostka, S.; Laubersheimer, A.; Dorken, B.; Wittmann-Liebold, B.; Bommert, K.; Otto, A. Identification of apoptosis-associated proteins in a human Burkitt lymphoma cell line. Cleavage of heterogeneous nuclear ribonucleoprotein A1 by caspase 3. J. Boil. Chem. 1998, 273, 28057–28064. [Google Scholar] [CrossRef]
- Shi, Y.; Frost, P.J.; Hoang, B.Q.; Benavides, A.; Sharma, S.; Gera, J.F.; Lichtenstein, A.K. IL-6-induced stimulation of c-myc translation in multiple myeloma cells is mediated by myc internal ribosome entry site function and the RNA-binding protein, hnRNP A1. Cancer Res. 2008, 68, 10215–10222. [Google Scholar] [CrossRef]
- Iervolino, A.; Santilli, G.; Trotta, R.; Guerzoni, C.; Cesi, V.; Bergamaschi, A.; Gambacorti-Passerini, C.; Calabretta, B.; Perrotti, D. hnRNP A1 nucleocytoplasmic shuttling activity is required for normal myelopoiesis and BCR/ABL leukemogenesis. Mol. Cell. Boil. 2002, 22, 2255–2266. [Google Scholar] [CrossRef]
- Zhang, S.; Wei, J.S.; Li, S.Q.; Badgett, T.C.; Song, Y.K.; Agarwal, S.; Coarfa, C.; Tolman, C.; Hurd, L.; Liao, H.; et al. MYCN controls an alternative RNA splicing program in high-risk metastatic neuroblastoma. Cancer Lett. 2016, 371, 214–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [PubMed]
- Barfeld, S.J.; Urbanucci, A.; Itkonen, H.M.; Fazli, L.; Hicks, J.L.; Thiede, B.; Rennie, P.S.; Yegnasubramanian, S.; DeMarzo, A.M.; Mills, I.G. c-Myc Antagonises the Transcriptional Activity of the Androgen Receptor in Prostate Cancer Affecting Key Gene Networks. EBioMedicine 2017, 18, 83–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyatt, A.W.; Gleave, M.E. Targeting the adaptive molecular landscape of castration-resistant prostate cancer. EMBO Mol. Med. 2015, 7, 878–894. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chan, S.C.; Brand, L.J.; Hwang, T.H.; Silverstein, K.A.; Dehm, S.M. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013, 73, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Loriot, Y.; Beraldi, E.; Zhang, F.; Wyatt, A.W.; Al Nakouzi, N.; Mo, F.; Zhou, T.; Kim, Y.; Monia, B.P.; et al. Generation 2.5 antisense oligonucleotides targeting the androgen receptor and its splice variants suppress enzalutamide-resistant prostate cancer cell growth. Clin. Cancer Res. 2015, 21, 1675–1687. [Google Scholar] [CrossRef]
- Mills, I.G. Maintaining and reprogramming genomic androgen receptor activity in prostate cancer. Nat. Rev. Cancer 2014, 14, 187–198. [Google Scholar] [CrossRef]
- Carabet, L.A.; Rennie, P.S.; Cherkasov, A. Therapeutic Inhibition of Myc in Cancer. Structural Bases and Computer-Aided Drug Discovery Approaches. Int. J. Mol. Sci. 2018, 20, 120. [Google Scholar] [CrossRef]
- Cao, S.; Zhan, Y.; Dong, Y. Emerging data on androgen receptor splice variants in prostate cancer. Endocr.-Relat. Cancer 2016, 23, T199–T210. [Google Scholar] [CrossRef] [Green Version]
- Koh, C.M.; Sabo, A.; Guccione, E. Targeting MYC in cancer therapy: RNA processing offers new opportunities. BioEssays 2016, 38, 266–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, R.; Lu, C.; Mostaghel, E.A.; Yegnasubramanian, S.; Gurel, M.; Tannahill, C.; Edwards, J.; Isaacs, W.B.; Nelson, P.S.; Bluemn, E.; et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012, 72, 3457–3462. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Indran, I.R.; Zhang, Z.W.; Tan, M.H.; Li, Y.; Lim, Z.L.; Hua, R.; Yang, C.; Soon, F.F.; Li, J.; et al. A novel prostate cancer therapeutic strategy using icaritin-activated arylhydrocarbon-receptor to co-target androgen receptor and its splice variants. Carcinogenesis 2015, 36, 757–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Babic, I.; Anderson, E.S.; Tanaka, K.; Guo, D.; Masui, K.; Li, B.; Zhu, S.; Gu, Y.; Villa, G.R.; Akhavan, D.; et al. EGFR mutation-induced alternative splicing of Max contributes to growth of glycolytic tumors in brain cancer. Cell Metab. 2013, 17, 1000–1008. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, M.; Tomonaga, T.; Matunis, M.; Avigan, M.; Krutzsch, H.; Dreyfuss, G.; Levens, D. Specific binding of heterogeneous ribonucleoprotein particle protein K to the human c-myc promoter, in vitro. J. Boil. Chem. 1993, 268, 18249–18258. [Google Scholar]
- Cogoi, S.; Rapozzi, V.; Cauci, S.; Xodo, L.E. Critical role of hnRNP A1 in activating KRAS transcription in pancreatic cancer cells: A molecular mechanism involving G4 DNA. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1389–1398. [Google Scholar] [CrossRef]
- Jo, O.D.; Martin, J.; Bernath, A.; Masri, J.; Lichtenstein, A.; Gera, J. Heterogeneous nuclear ribonucleoprotein A1 regulates cyclin D1 and c-myc internal ribosome entry site function through Akt signaling. J. Boil. Chem. 2008, 283, 23274–23287. [Google Scholar] [CrossRef]
- Jain, N.; Lin, H.C.; Morgan, C.E.; Harris, M.E.; Tolbert, B.S. Rules of RNA specificity of hnRNP A1 revealed by global and quantitative analysis of its affinity distribution. Proc. Natl. Acad. Sci. USA 2017, 114, 2206–2211. [Google Scholar] [CrossRef] [Green Version]
- Howard, J.M.; Lin, H.; Wallace, A.J.; Kim, G.; Draper, J.M.; Haeussler, M.; Katzman, S.; Toloue, M.; Liu, Y.; Sanford, J.R. HNRNPA1 promotes recognition of splice site decoys by U2AF2 in vivo. Genome Res. 2018, 28, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Tavanez, J.P.; Madl, T.; Kooshapur, H.; Sattler, M.; Valcarcel, J. hnRNP A1 proofreads 3′ splice site recognition by U2AF. Mol. Cell 2012, 45, 314–329. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Xie, N.; Sun, S.; Plymate, S.; Mostaghel, E.; Dong, X. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene 2014, 33, 3140–3150. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.E.; Meagher, J.L.; Levengood, J.D.; Delproposto, J.; Rollins, C.; Stuckey, J.A.; Tolbert, B.S. The First Crystal Structure of the UP1 Domain of hnRNP A1 Bound to RNA Reveals a New Look for an Old RNA Binding Protein. J. Mol. Boil. 2015, 427, 3241–3257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, N.; Chen, Y.; Mitchell, S.H.; Young, C.Y. Quercetin inhibits the expression and function of the androgen receptor in LNCaP prostate cancer cells. Carcinogenesis 2001, 22, 409–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, D.; Guo, X.; Zhang, E.; Zi, F.; Chen, J.; Chen, Q.; Lin, X.; Yang, L.; Li, Y.; Wu, W.; et al. Quercetin induces cell apoptosis of myeloma and displays a synergistic effect with dexamethasone in vitro and in vivo xenograft models. Oncotarget 2016, 7, 45489–45499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granato, M.; Rizzello, C.; Romeo, M.A.; Yadav, S.; Santarelli, R.; D’Orazi, G.; Faggioni, A.; Cirone, M. Concomitant reduction of c-Myc expression and PI3K/AKT/mTOR signaling by quercetin induces a strong cytotoxic effect against Burkitt’s lymphoma. Int. J. Biochem. Cell Boil. 2016, 79, 393–400. [Google Scholar] [CrossRef]
- Ko, C.C.; Chen, Y.J.; Chen, C.T.; Liu, Y.C.; Cheng, F.C.; Hsu, K.C.; Chow, L.P. Chemical proteomics identifies heterogeneous nuclear ribonucleoprotein (hnRNP) A1 as the molecular target of quercetin in its anti-cancer effects in PC-3 cells. J. Boil. Chem. 2014, 289, 22078–22089. [Google Scholar] [CrossRef]
- Tummala, R.; Lou, W.; Gao, A.C.; Nadiminty, N. Quercetin Targets hnRNPA1 to Overcome Enzalutamide Resistance in Prostate Cancer Cells. Mol. Cancer Ther. 2017, 16, 2770–2779. [Google Scholar] [CrossRef]
- Rollins, C.; Levengood, J.D.; Rife, B.D.; Salemi, M.; Tolbert, B.S. Thermodynamic and phylogenetic insights into hnRNP A1 recognition of the HIV-1 exon splicing silencer 3 element. Biochemistry 2014, 53, 2172–2184. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), 2013. 08; Chemical Computing Group ULC: Montreal, QC, Canada, 2018.
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Scoring Analysis Tool for Non-Bonded Intermolecular Interactions: H-Bonds, Transition Metal, Water Bridges, Hydrophobic; Scientific Vector Language (SVL) Source Code; Chemical Computing Group ULC: Montreal, QC, Canada, 2018.
- Neves, M.A.; Totrov, M.; Abagyan, R. Docking and scoring with ICM: The benchmarking results and strategies for improvement. J. Comput.-Aided Mol. Des. 2012, 26, 675–686. [Google Scholar] [CrossRef] [PubMed]
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput.-Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGovern, S.L.; Shoichet, B.K. Kinase inhibitors: Not just for kinases anymore. J. Med. Chem. 2003, 46, 1478–1483. [Google Scholar] [CrossRef]
- Baell, J.; Walters, M.A. Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef] [Green Version]
- ZINC3869685. Available online: http://zinc15.docking.org/substances/ZINC000003869685/ (accessed on 6 February 2019).
- FAFDrugs4 Home. Available online: http://fafdrugs4.mti.univ-paris-diderot.fr/ (accessed on 6 February 2019).
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FAF-QED—FAFDrugs4. Available online: http://fafdrugs4.mti.univ-paris-diderot.fr/fafqed.html (accessed on 6 February 2019).
- Maestro, 2018-4; Schrödinger LLC: New York, NY, USA, 2018.
- OpenEye Scientific Software, Santa Fe, NM. Available online: http://www.eyesopen.com (accessed on 6 February 2019).
- mol_rmsd Calculate RMSD’s for Docking Results; Scientific Vector Language (SVL) Source Code; Chemical Computing Group ULC: Montreal, QC, Canada, 2018.
- Prime, 2018-4; Schrödinger LLC: New York, NY, USA, 2018.
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmond, 2018-4; Schrödinger LLC: New York, NY, USA, 2018.
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Tuckerman, M.; Berne, B.J.; Martyna, G.J. Reversible multiple time scale molecular dynamics. J. Chem. Phys. 1992, 97, 1990–2001. [Google Scholar] [CrossRef]
- Carabet, L.A.; Lallous, N.; Leblanc, E.; Ban, F.; Morin, H.; Lawn, S.; Ghaidi, F.; Lee, J.; Mills, I.G.; Gleave, M.E.; et al. Computer-aided drug discovery of Myc-Max inhibitors as potential therapeutics for prostate cancer. Eur. J. Med. Chem. 2018, 160, 108–119. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carabet, L.A.; Leblanc, E.; Lallous, N.; Morin, H.; Ghaidi, F.; Lee, J.; Rennie, P.S.; Cherkasov, A. Computer-Aided Discovery of Small Molecules Targeting the RNA Splicing Activity of hnRNP A1 in Castration-Resistant Prostate Cancer. Molecules 2019, 24, 763. https://doi.org/10.3390/molecules24040763
Carabet LA, Leblanc E, Lallous N, Morin H, Ghaidi F, Lee J, Rennie PS, Cherkasov A. Computer-Aided Discovery of Small Molecules Targeting the RNA Splicing Activity of hnRNP A1 in Castration-Resistant Prostate Cancer. Molecules. 2019; 24(4):763. https://doi.org/10.3390/molecules24040763
Chicago/Turabian StyleCarabet, Lavinia A., Eric Leblanc, Nada Lallous, Helene Morin, Fariba Ghaidi, Joseph Lee, Paul S. Rennie, and Artem Cherkasov. 2019. "Computer-Aided Discovery of Small Molecules Targeting the RNA Splicing Activity of hnRNP A1 in Castration-Resistant Prostate Cancer" Molecules 24, no. 4: 763. https://doi.org/10.3390/molecules24040763