Machine Learning Approach for Determining the Formation of β-Lactam Antibiotic Complexes with Cyclodextrins Using Multispectral Analysis



Abstract
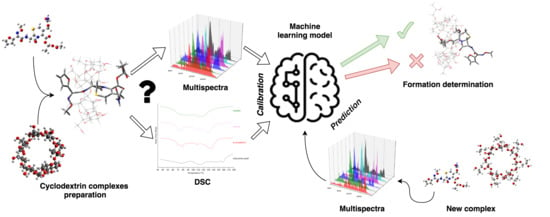
:
1. Introduction
2. Results and Discussion
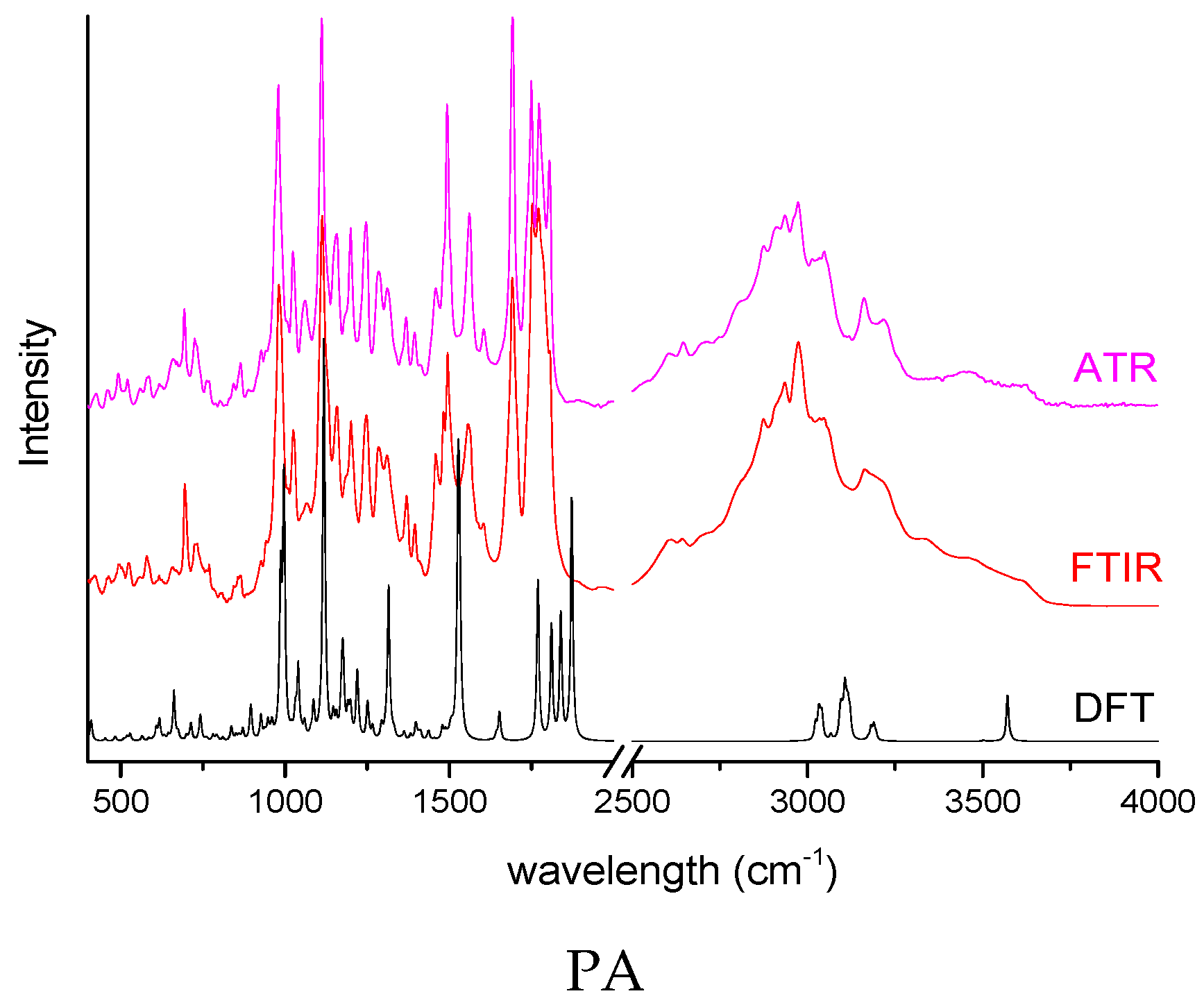
2.1. Determination of Complexes Formation
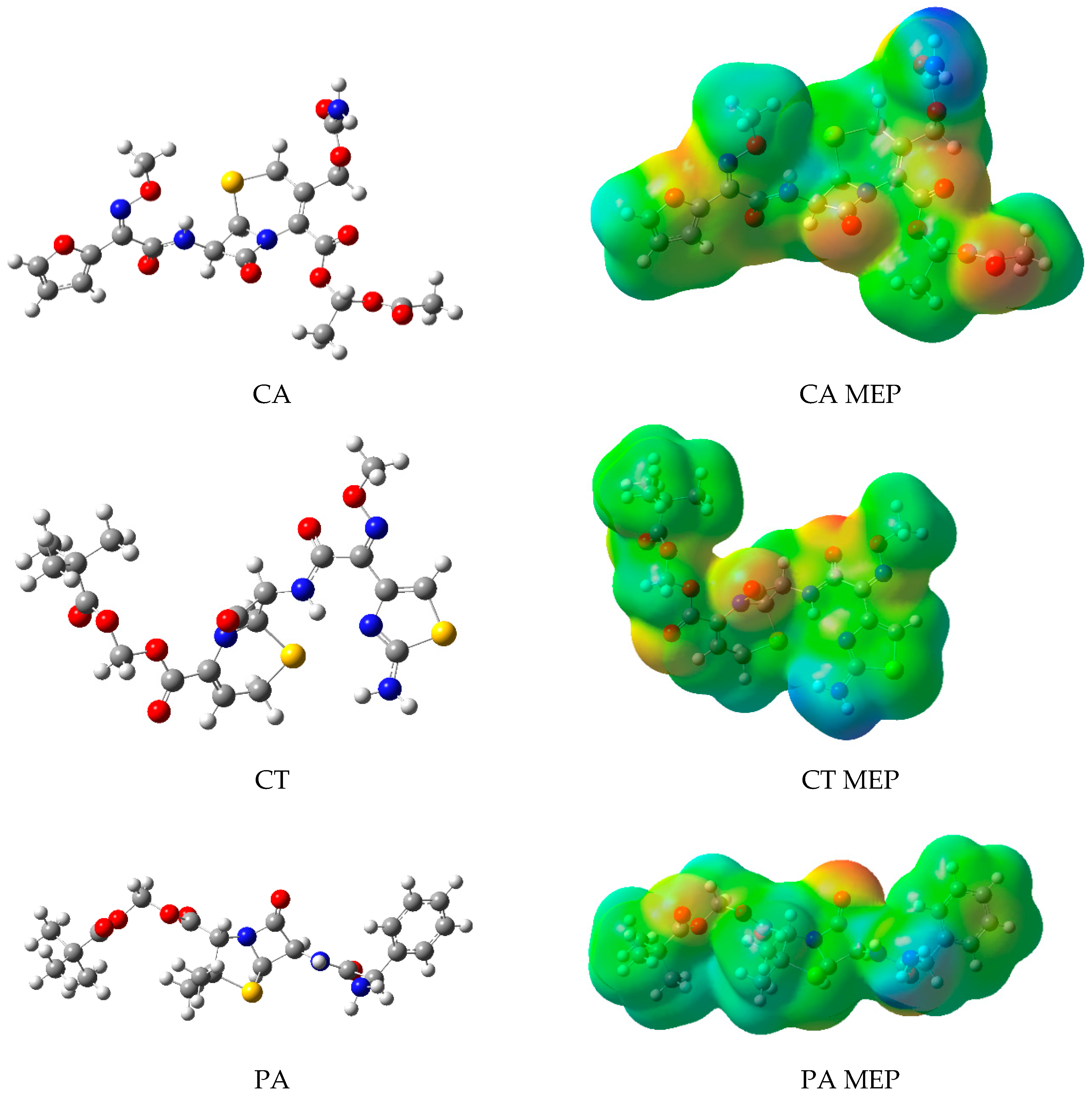
2.2. Molecular Modeling
2.3. Machine Learning
2.4. Discussion
3. Materials and Methods
3.1. Materials
3.2. Instrumentation
3.3. Computation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paczkowska, M.; Mizera, M.; Szymanowska-Powałowska, D.; Lewandowska, K.; Błaszczak, W.; Gościańska, J.; Cielecka-Piontek, J. β-Cyclodextrin complexation as an effective drug delivery system for meropenem. Eur. J. Pharm. Biopharm. 2016, 99, 24–34. [Google Scholar] [CrossRef]
- Paczkowska, M.; Szymanowska-Powałowska, D.; Mizera, M.; Siąkowska, D.; Błaszczak, W.; Piotrowska-Kempisty, H.; Cielecka-Piontek, J. Cyclodextrins as multifunctional excipients: Influence of inclusion into β-cyclodextrin on physicochemical and biological properties of tebipenem pivoxil. PLoS ONE 2019, 14, e0210694. [Google Scholar] [CrossRef]
- Aljuffali, I.A.; Lin, C.F.; Chen, C.H.; Fang, J.Y. The codrug approach for facilitating drug delivery and bioactivity. Expert Opin. Drug Deliv. 2016, 13, 1311–1325. [Google Scholar] [CrossRef]
- Jornada, D.H.; dos Santos Fernandes, G.F.; Chiba, D.E.; de Melo, T.R.F.; dos Santos, J.L.; Chung, M.C. The prodrug approach: A successful tool for improving drug solubility. Molecules 2015, 21, 42. [Google Scholar] [CrossRef]
- Gupta, D.; Gupta, S.V.; Lee, K.D.; Amidon, G.L. Chemical and enzymatic stability of amino acid prodrugs containing methoxy, ethoxy and propylene glycol linkers. Mol. Pharm. 2009, 6, 1604–1611. [Google Scholar] [CrossRef]
- Karaman, R. Prodrugs for Masking the Bitter Taste of Drugs. In Application of Nanotechnology in Drug Delivery; InTech: London, UK, 2014; pp. 399–445. [Google Scholar]
- Liu, Y.; Zhao, D.; Sun, M.; Wei, W.; Wang, Y.; Zhou, J.; Kan, Q. Covalently mucoadhesive amphiphilic prodrug of 5-fluorouracil for enhanced permeation and improved oral absorption. Drug Deliv. Transl. Res. 2018, 8, 645–656. [Google Scholar] [CrossRef]
- Placzek, A.T.; Ferrara, S.J.; Hartley, M.D.; Sanford-Crane, H.S.; Meinig, J.M.; Scanlan, T.S. Sobetirome prodrug esters with enhanced blood–brain barrier permeability. Bioorgan. Med. Chem. 2016, 24, 5842–5854. [Google Scholar] [CrossRef] [Green Version]
- Leifer, F.; Omiatek, D.; Malinin, V.; Ong, J.; Li, Z.; Klecha, P.; Perkins, W. Prolonged activity of inhaled treprostinil prodrug nanoparticles in a rat model of pulmonary arterial hypertension. Eur. Respir. J. 2014, 44, P2356. [Google Scholar]
- Chu, K.S.; Finniss, M.C.; Schorzman, A.N.; Kuijer, J.L.; Luft, J.C.; Bowerman, C.J.; DeSimone, J.M. Particle replication in nonwetting templates nanoparticles with tumor selective alkyl silyl ether docetaxel prodrug reduces toxicity. Nano Lett. 2014, 14, 1472–1476. [Google Scholar] [CrossRef]
- Li, S.L.; Hou, Y.; Hu, Y.; Yu, J.; Wei, W.; Lu, H. Phosphatase-triggered cell-selective release of a Pt (IV)-backboned prodrug-like polymer for an improved therapeutic index. Biomater. Sci. 2017, 5, 1558–1566. [Google Scholar] [CrossRef]
- Beaumont, K.; Webster, R.; Gardner, I.; Dack, K. Design of ester prodrugs to enhance oral absorption of poorly permeable compounds: Challenges to the discovery scientist. Curr. Drug Metab. 2003, 4, 461–485. [Google Scholar] [CrossRef]
- Soares, F.L.; Carneiro, R.L. Green synthesis of ibuprofen–nicotinamide cocrystals and in-line evaluation by Raman spectroscopy. Cryst. Growth Design 2013, 13, 1510–1517. [Google Scholar] [CrossRef]
- Trasi, N.S.; Taylor, L.S. Dissolution performance of binary amorphous drug combinations—Impact of a second drug on the maximum achievable supersaturation. Int. J. Pharm. 2015, 496, 282–290. [Google Scholar] [CrossRef]
- Ige, P.P.; Baria, R.K.; Gattani, S.G. Fabrication of fenofibrate nanocrystals by probe sonication method for enhancement of dissolution rate and oral bioavailability. Colloids Surf. B 2013, 108, 366–373. [Google Scholar] [CrossRef]
- Penkina, A.; Semjonov, K.; Hakola, M.; Vuorinen, S.; Repo, T.; Yliruusi, J.; Heinämäki, J. Towards improved solubility of poorly water-soluble drugs: Cryogenic co-grinding of piroxicam with carrier polymers. Drug Dev. Ind. Pharm. 2015, 42, 378–388. [Google Scholar] [CrossRef]
- Cappello, B.; Di Maio, C.L.; Iervolino, M.; Miro, A. Improvement of solubility and stability of valsartan by hydroxypropyl-beta-cyclodextrin. J. Inclus. Phenom. Macrocycl. Chem. 2006, 54, 289–294. [Google Scholar] [CrossRef]
- Jambhekar, S.S.; Breen, P. Cyclodextrins in pharmaceutical formulations I: Structure and physicochemical properties, formation of complexes, and types of complex. Drug Discov. Today 2016, 21, 356–362. [Google Scholar] [CrossRef]
- Miller, L.A.; Carrier, R.L.; Ahmed, I. Practical considerations in development of solid dosage forms that contain cyclodextrin. J. Pharm. Sci. 2007, 96, 1691–1707. [Google Scholar] [CrossRef]
- Ghosh, A.; Biswas, S.; Ghosh, T. Preparation and evaluation of silymarin β-cyclodextrin molecular inclusion complexes. J. Young Pharm. 2011, 3, 205–210. [Google Scholar] [CrossRef]
- Marzouk, M.A.; Kassem, A.A.; Samy, A.M.; Amer, R.I. Comparative evaluation of ketoconazole-β-cyclodextrin systems prepared by coprecipitation and kneading. Drug Discov. Ther. 2010, 4, 380–387. [Google Scholar]
- Mohan, P.K.; Sreelakshmi, G.; Muraleedharan, C.V.; Joseph, R. Water soluble complexes of curcumin with cyclodextrins: Characterization by FT-Raman spectroscopy. Vib. Spectr. 2012, 62, 77–84. [Google Scholar] [CrossRef]
- Aigner, Z.; Hassan, H.B.; Berkesi, O.; Kata, M.; Erős, I. Thermoanalytical, FTIR and X-ray studies of gemfibrozil-cyclodextrin complexes. J. Therm. Anal. Calorimetry 2005, 81, 267–272. [Google Scholar] [CrossRef]
- Mura, P. Analytical techniques for characterization of cyclodextrin complexes in the solid state: A review. J. Pharm. Biomed. Anal. 2015, 113, 226–238. [Google Scholar] [CrossRef]
- Ellis, L.D.; Buteau, S.; Hames, S.G.; Thompson, L.M.; Hall, D.S.; Dahn, J.R. A New Method for Determining the Concentration of Electrolyte Components in Lithium-Ion Cells, Using Fourier Transform Infrared Spectroscopy and Machine Learning. J. Electrochem. Soc. 2018, 165, A256–A262. [Google Scholar] [CrossRef] [Green Version]
- Nalla, R.; Pinge, R.; Narwaria, M.; Chaudhury, B. Priority based functional group identification of organic molecules using machine learning. In Proceedings of the ACM India Joint International Conference on Data Science and Management of Data; ACM: New York, NY, USA, 2018; pp. 201–209. [Google Scholar]
- Toplak, M.; Birarda, G.; Read, S.; Sandt, C.; Rosendahl, S.M.; Vaccari, L.; Borondics, F. Infrared Orange: Connecting Hyperspectral Data with Machine Learning. Synchrotron Radiat. News 2017, 30, 40–45. [Google Scholar] [CrossRef]
- Kharyuk, P.; Nazarenko, D.; Oseledets, I. Comparative study of Discrete Wavelet Transforms and Wavelet Tensor Train decomposition to feature extraction of FTIR data of medicinal plants. arXiv, 2018; arXiv:1807.07099. [Google Scholar]
- Zhao, Y.; Zhang, C.; Zhu, S.; Gao, P.; Feng, L.; He, Y. Non-destructive and rapid variety discrimination and visualization of single grape seed using near-infrared hyperspectral imaging technique and multivariate analysis. Molecules 2018, 23, 1352. [Google Scholar] [CrossRef]
- Shah, M.; Pore, Y.; Dhawale, S.; Burade, K.; Kuchekar, B. Physicochemical characterization of spray dried ternary micro-complexes of cefuroxime axetil with hydroxypropyl-β-cyclodextrin. J. Inclus. Phenom. Macrocycl. Chem. 2013, 76, 391–401. [Google Scholar] [CrossRef]
- Sapte, S.; Pore, Y. Inclusion complexes of cefuroxime axetil with β-cyclodextrin: Physicochemical characterization, molecular modeling and effect of L-Arginine on complexation. J. Pharm. Anal. 2016, 6, 300–306. [Google Scholar] [CrossRef]
- Puliti, R.; Mattia, C.A.; Paduano, L. Crystal structure of a new α-cyclodextrin hydrate form. Molecular geometry and packing features: Disordered solvent contribution. Carbohydr. Res. 1998, 310, 1–8. [Google Scholar] [CrossRef]
- Ramos, A.I.; Braga, T.M.; Silva, P.; Fernandes, J.A.; Ribeiro-Claro, P.; Lopes, M.D.F.S.; Braga, S.S. Chloramphenicol cyclodextrin inclusion compounds: Co-dissolution and mechanochemical preparations and antibacterial action. CrystEngComm 2013, 15, 2822–2834. [Google Scholar] [CrossRef]
- Harata, K. Crystal structure of γ-cyclodextrin at room temperature. Chem. Lett. 1984, 13, 641–644. [Google Scholar] [CrossRef]
- O'Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Stewart Computational Chemistry. Stewart, Stewart Computational Chemistry; Stewart Computational Chemistry: Colorado Springs, CO, USA, 2007. [Google Scholar]
- Olson, R.S.; Urbanowicz, R.J.; Andrews, P.C.; Lavender, N.A.; Moore, J.H. Automating biomedical data science through tree-based pipeline optimization. Appl. Evol. Comput. 2016, 123–137. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python; JMLR 12; JMLR, Inc. and Microtome Publishing: USA, 2011; pp. 2825–2830. [Google Scholar]
Sample Availability: Samples of the CA, CT and PA complexes with CDs are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CA | CT | PA | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Cyclodextrin | Physical Mixture | Complex | Change | Physical Mixture | Complex | Change | Physical Mixture | Complex | Change |
| αCD | 77 °C 138 °C | - - | Intensity ↓ Intensity ↓ | 71 °C 126 °C 164 °C | - - - | Intensity ↓ Intensity ↓ Intensity ↓ | 76 °C 131 °C | - - | Intensity ↓ Intensity ↓ |
| βCD | 114 °C | 116 °C | Peak ↔ Intensity ↓ | 113 °C 164 °C | - - | Intensity ↓ Intensity ↓ | 120 °C | - | Intensity ↓ |
| γCD | 100 °C | 103 °C | Peak ↔ Intensity ↓ | 86 °C 164 °C | 100 °C - | Peak ↔ Intensity ↑ Intensity ↓ | 114 °C | 100 °C | Peak ↔ Intensity ↓ |
| HPαCD | - | - | No changes | 76 °C 164 °C | 93 °C - | Peak ↔, Intensity ↑ Intensity ↓ | 84 °C 138 °C | 84 °C 140 °C | Intensity ↑ Peak ↔ Intensity ↓ |
| HPβCD | 83 °C | 87 °C | Peak ↔ Intensity ↓ | 83 °C 164 °C | 91 °C 164 °C | Intensity ↓ Intensity ↓ | 84 °C 138 °C | 100 °C - | Peak ↔ Intensity ↑ Intensity ↓ |
| HPγCD | 91 °C | 85 °C | Peak ↔ Intensity ↓ | 85 °C 164 °C | 95 °C 164 °C | Intensity ↑ Intensity ↓ | 84 °C 138 °C | 94 °C 145 °C | Peak ↔ Intensity ↓ Peak ↔ Intensity ↓ |
| MβCD | 89 °C | 92 °C | Peak ↔ Intensity ↓ | 93 °C 165 °C | 93 °C 165 °C | Intensity ↑ Intensity ↓ | 82 °C 138 °C | 92 °C 145 °C | Peak ↔ Intensity ↑ Peak ↔ Intensity ↓ |
| Cefuroxime axetil | Cefetamet pivoxil | Pivamipicillin | ||||
|---|---|---|---|---|---|---|
| Simulation Favored | Experimentally Favored | Simulation Favored | Experimentally Favored | Simulation Favored | Experimentally Favored | |
| α | −157.31 | −134.83 | −239.54 | −235.75 | −197.79 | −149.85 |
| β | −238.22 | −145.17 | −279.69 | −131.33 | −263.39 | −195.71 |
| γ | −255.57 | −196.71 | −291.79 | −178.73 | −243.19 | −125.94 |
| HPα | 486.92 | 558.60 | −250.64 | −182.03 | −235.03 | −180.66 |
| Mβ | −256.15 | −168.48 | −185.90 | −144.22 | −236.37 | −173.36 |
| HPβ | −904.60 | 352.80 | −268.65 | −259.74 | −193.11 | −223.94 |
| HPγ | −352.92 | −230.65 | −282.78 | −263.54 | −246.71 | −293.35 |
| CA | CT | PA | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Complexed | Physical mixtures | Complexed | Physical mixtures | Complexed | Physical mixtures | |||||||
| Predicted | True value | Predicted | True value | Predicted | True value | Predicted | True value | Predicted | True value | Predicted | True value | |
| αCD | True | True | True | False | True | True | False | False | True | True | False | False |
| βCD | True | True | False | False | True | True | False | False | True | True | False | False |
| γCD | True | True | False | False | True | True | False | False | True | True | False | False |
| HPαCD | False | False | False | False | True | True | False | False | True | True | False | False |
| HPβCD | False | True | False | False | True | True | False | False | True | True | False | False |
| HPγCD | True | True | False | False | True | True | True | False | True | True | False | False |
| MβCD | True | True | False | False | False | True | False | False | True | True | False | False |
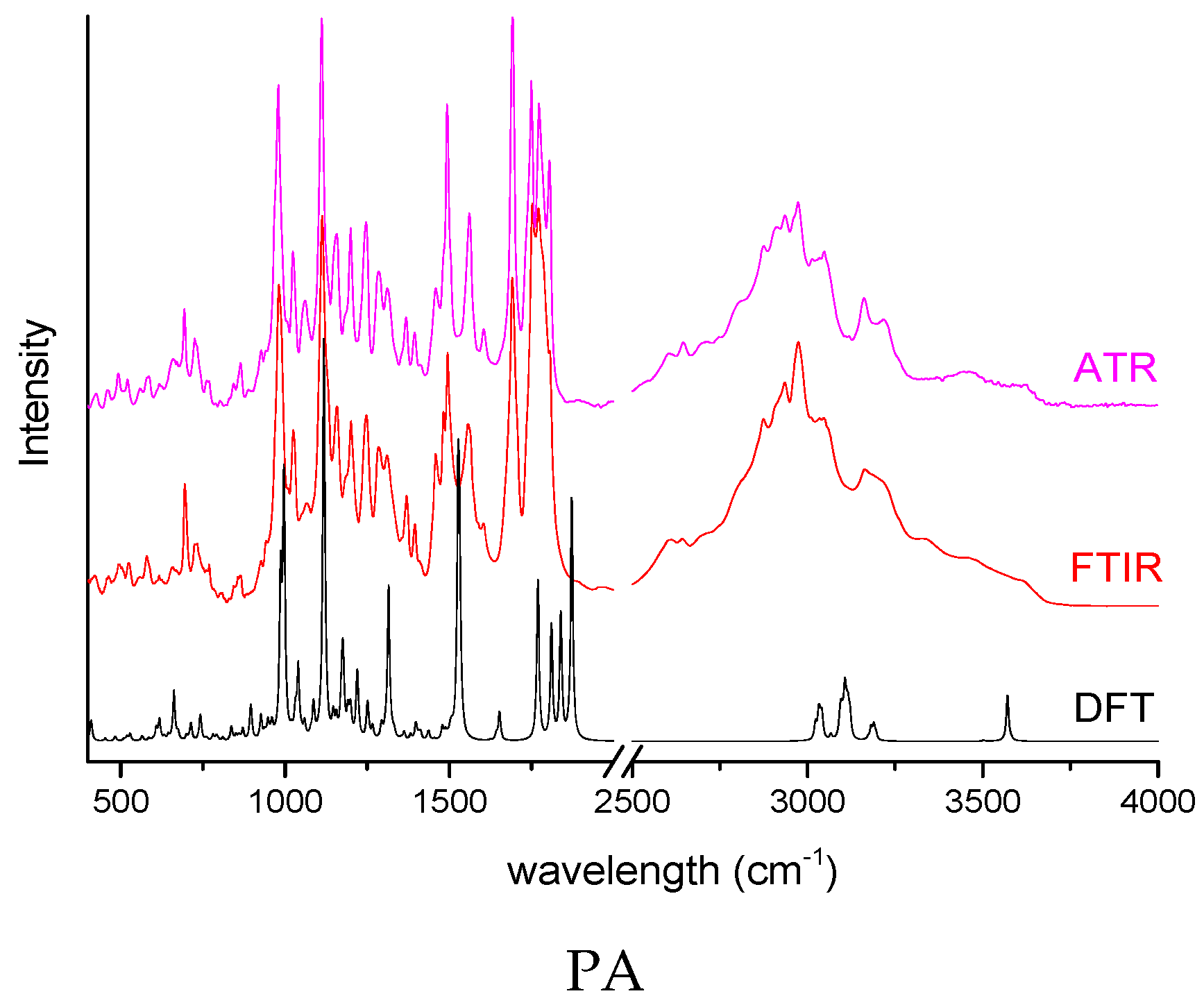
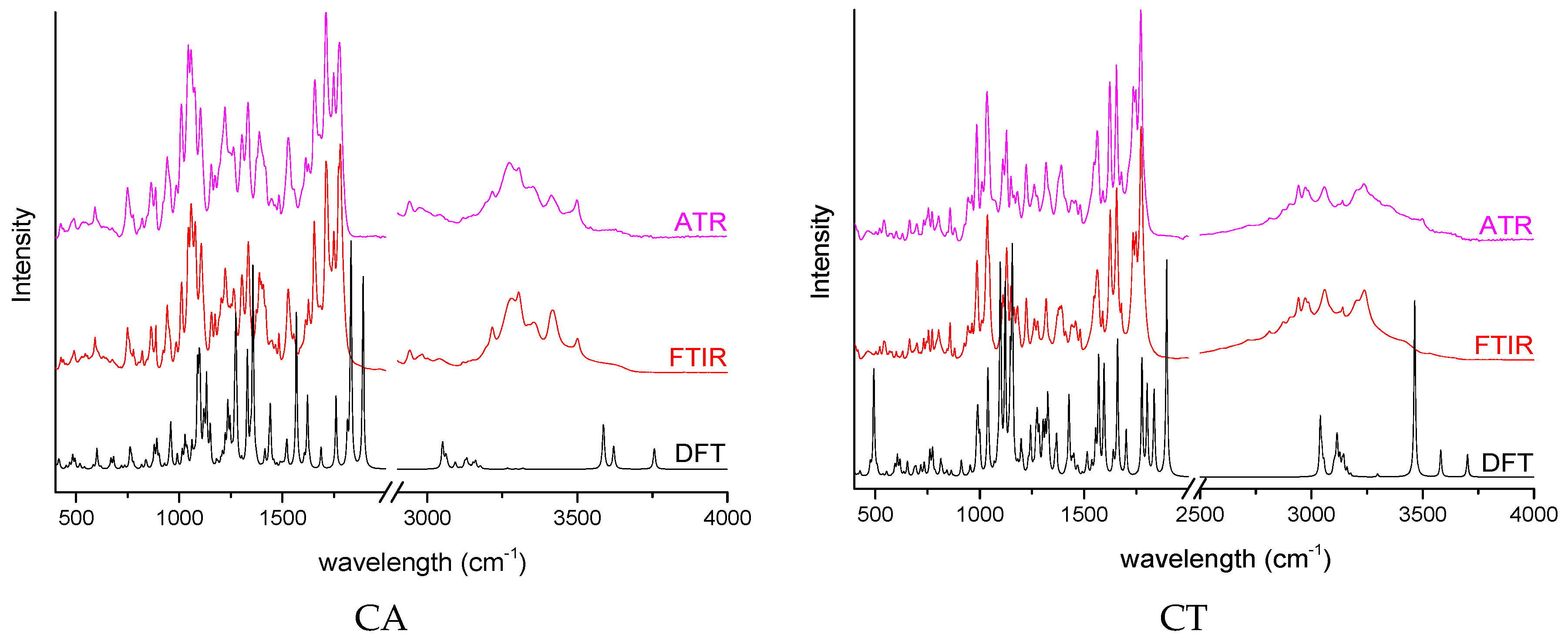
| API | Peak [cm−1] | Theoretical | Description |
|---|---|---|---|
| CA | 1772 | 1832 | C=O s in (acetyloxy)ethyl group C=O s in ((aminocarbonyl) oxy)methyl group |
| 1264 | 1274 | C–O s in (acetyloxy)ethyl group C–H w in ((aminocarbonyl) oxy)methyl group | |
| CT | 1774 | 1838 | C=O s in pivoxil group |
| 1458 | 1479 | CH2 sc in pivoxil group | |
| 1277 | 1275 | C-C s between the thizaol ring and cephem group C-N-H b-ip between the thizaol ring and cephem group | |
| PA | 1774 | 1838 | C=O s in pivoxil group |
| 1458 | 1479 | CH2 sc in pivoxil group | |
| 1371 | 1362 | C-H b in β-lactam ring | |
| 1283 | 1293 | C-C s in pivoxil group |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizera, M.; Lewandowska, K.; Miklaszewski, A.; Cielecka-Piontek, J. Machine Learning Approach for Determining the Formation of β-Lactam Antibiotic Complexes with Cyclodextrins Using Multispectral Analysis. Molecules 2019, 24, 743. https://doi.org/10.3390/molecules24040743
Mizera M, Lewandowska K, Miklaszewski A, Cielecka-Piontek J. Machine Learning Approach for Determining the Formation of β-Lactam Antibiotic Complexes with Cyclodextrins Using Multispectral Analysis. Molecules. 2019; 24(4):743. https://doi.org/10.3390/molecules24040743
Chicago/Turabian StyleMizera, Mikołaj, Kornelia Lewandowska, Andrzej Miklaszewski, and Judyta Cielecka-Piontek. 2019. "Machine Learning Approach for Determining the Formation of β-Lactam Antibiotic Complexes with Cyclodextrins Using Multispectral Analysis" Molecules 24, no. 4: 743. https://doi.org/10.3390/molecules24040743