Synthesis of a Conformationally Stable Atropisomeric Pair of Biphenyl Scaffold Containing Additional Stereogenic Centers

 , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Methods
3.2. X-ray Crystallography
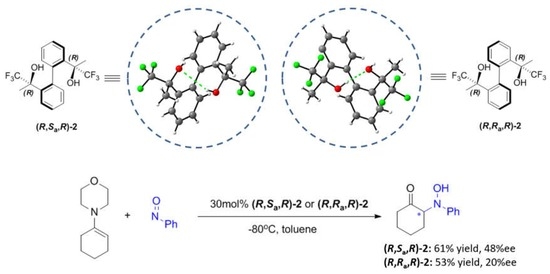
3.3. Procedure for Synthesis of (R,Sa,R)-2 and (R, Ra,R)-2
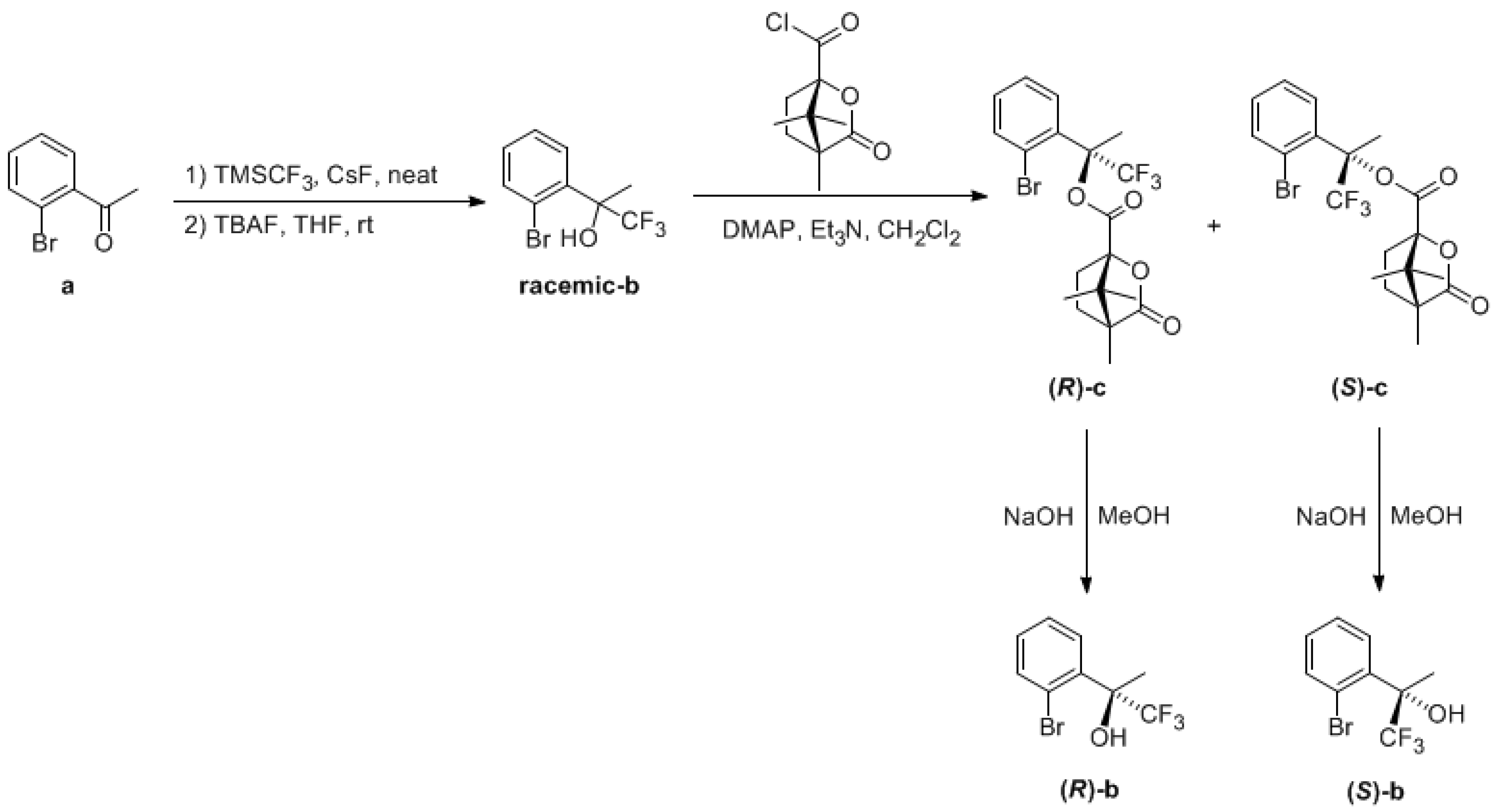
3.3.1. Synthesis of Racemic 2-(2-bromophenyl)-1,1,1-trifluoropropan-2-ol, racemic-b
3.3.2. Synthesis of ((1S,4R)-((R)-2-(2-bromophenyl)-1,1,1-trifluoropropan-2-yl) 4,7,7-trimethyl-3-oxo-2-oxabicyclo (2.2.1)heptane-1-carboxylate), (R)-c, and ((1S,4R)-((S)-2-(2-bromophenyl)-1,1,1-trifluoropropan-2-yl) 4,7,7-trimethyl-3-oxo-2-oxabicyclo (2.2.1)heptane-1-carboxylate), (R)-c
3.3.3. Synthesis of (R)-2-(2-bromophenyl)-1,1,1-trifluoropropan-2-ol, (R)-b
3.3.4. Synthesis of (Sa)-(2R,2′′R)-((1,1′-biphenyl)-2,2′-diyl)bis(1,1,1-trifluoropropan-2-ol) (R,Sa,R)-2 and (Ra)-(2R,2′R)-((1,1′-biphenyl)-2,2′-diyl)bis(1,1,1-trifluoropropan-2-ol) (R,Ra,R)-2
3.4. General Procedure for Asymmetric N-Nitroso Aldol Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Janey, J.M. Recent advances in catalytic, enantioselective alpha aminations and alpha oxygenations of carbonyl compounds. Angew. Chem. Int. Ed. 2005, 44, 4292–4300. [Google Scholar] [CrossRef] [PubMed]
- Baidya, M.; Griffin, K.A.; Yamamoto, H. Catalytic Enantioselective O-Nitrosocarbonyl Aldol Reaction of beta-Dicarbonyl Compounds. J. Am. Chem. Soc. 2012, 134, 18566–18569. [Google Scholar] [CrossRef] [PubMed]
- Merino, P.; Tejero, T. Organocatalyzed asymmetric alpha-aminoxylation of aldehydes and ketones—An efficient access to enantiomerically pure alpha-hydroxycarbonyl compounds, diols, and even amino alcohols. Angew. Chem. Int. Ed. 2004, 43, 2995–2997. [Google Scholar] [CrossRef] [PubMed]
- Merino, P.; Tejero, T.; Delso, I.; Matute, R. Recent Advances on Asymmetric Nitroso Aldol Reaction. Synthesis (stuttg) 2016, 48, 653–676. [Google Scholar] [CrossRef] [Green Version]
- Matsuzawa, M.; Kakeya, H.; Yamaguchi, J.; Shoji, M.; Onose, R.; Osada, H.; Hayashi, Y. Enantio- and diastereoselective total synthesis of (+)-panepophenanthrin, a ubiquitin-activating enzyme inhibitor, and biological properties of its new derivatives. Chem. Asian J. 2006, 1, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Momiyama, N.; Yamamoto, H. Simple synthesis of alpha-hydroxyamino carbonyl compounds: New scope of the nitroso aldol reaction. Org. Lett. 2002, 4, 3579–3582. [Google Scholar] [CrossRef] [PubMed]
- Momiyama, N.; Yamamoto, H. Catalytic enantioselective synthesis of alpha-aminooxy and alpha-hydroxy ketone using nitrosobenzene. J. Am. Chem. Soc. 2003, 125, 6038–6039. [Google Scholar] [CrossRef] [PubMed]
- Momiyama, N.; Yamamoto, H. Lewis acid promoted, O-selective, nucleophilic addition of silyl enol ethers to N=O bonds. Angew. Chem. Int. Ed. 2002, 41, 2986–2988. [Google Scholar] [CrossRef]
- Zhong, G.F. A facile and rapid route to highly enantiopure 1,2-diols by novel catalytic asymmetric alpha-aminoxylation of aldehydes. Angew. Chem. Int. Ed. 2003, 42, 4247–4250. [Google Scholar] [CrossRef]
- Brown, S.P.; Brochu, M.P.; Sinz, C.J.; MacMillan, D.W.C. The direct and enantioselective organocatalytic alpha-oxidation of aldehydes. J. Am. Chem. Soc. 2003, 125, 10808–10809. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Yamaguchi, J.; Hibino, K.; Shoji, M. Direct proline catalyzed asymmetric alpha-aminooxylation of aldehydes. Tetrahedron Lett. 2003, 44, 8293–8296. [Google Scholar] [CrossRef]
- Bogevig, A.; Sunden, H.; Cordova, A. Direct catalytic enantioselective alpha-aminoxylation of ketones: A stereoselective synthesis of alpha-hydroxy and alpha,alpha ‘-dihydroxy ketones. Angew. Chem. Int. Ed. 2004, 43, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.M.; Niu, H.Y.; Xue, M.X.; Guo, Q.X.; Cun, L.F.; Mi, A.Q.; Jiang, Y.Z.; Wang, J.J. L-Proline in an ionic liquid as an efficient and reusable catalyst for direct asymmetric alpha-aminoxylation of aldehydes and ketones. Green Chem. 2006, 8, 682–684. [Google Scholar] [CrossRef]
- Huang, K.; Huang, Z.Z.; Li, X.L. Highly enantioselective alpha-aminoxylation of aldehydes and ketones in ionic liquids. J. Org. Chem. 2006, 71, 8320–8323. [Google Scholar] [CrossRef]
- Poe, S.L.; Bogdan, A.R.; Mason, B.P.; Steinbacher, J.L.; Opalka, S.M.; McQuade, D.T. Use of Bifunctional Ureas to Increase the Rate of Proline-Catalyzed alpha-Aminoxylations. J. Org. Chem. 2009, 74, 1574–1580. [Google Scholar] [CrossRef] [PubMed]
- Demir, A.S.; Basceken, S. Self-assembly of an organocatalyst for the enantioselective synthesis of Michael adducts and alpha-aminoxy alcohols in a nonpolar medium. Tetrahedron: Asymmetry 2013, 24, 1218–1224. [Google Scholar] [CrossRef]
- Mailhol, D.; Castillo, J.C.; Mohanan, K.; Abonia, R.; Coquerel, Y.; Rodriguez, J. Practical and Efficient Organocatalytic Enantioselective alpha-Hydroxyamination Reactions of beta-Ketoamides. ChemCatChem 2013, 5, 1192–1199. [Google Scholar] [CrossRef]
- Maji, B.; Yamamoto, H. Proline-Tetrazole-Catalyzed Enantioselective N-Nitroso Aldol Reaction of Aldehydes with In Situ Generated Nitrosocarbonyl Compounds. Angew. Chem. Int. Ed. 2014, 53, 8714–8717. [Google Scholar] [CrossRef]
- Guo, H.M.; Cheng, L.; Cun, L.F.; Gong, L.Z.; Mi, A.Q.; Jiang, Y.Z. L-prolinamide-catalyzed direct nitroso aldol reactions of alpha-branched aldehydes: A distinct regioselectivity from that with L-proline. Chem. Commun. 2006, 429–431. [Google Scholar] [CrossRef]
- Kano, T.; Ueda, M.; Takai, J.; Maruoka, K. Direct asymmetric hydroxyamination reaction catalyzed by an axially chiral secondary amine catalyst. J. Am. Chem. Soc. 2006, 128, 6046–6047. [Google Scholar] [CrossRef]
- Momiyama, N.; Yamamoto, Y.; Yamamoto, H. Diastereo- and enantioselective synthesis of nitroso Diels-Alder-type bicycloketones using dienamine: Mechanistic insight into sequential nitroso aldol/Michael reaction and application for optically pure 1-amino-3,4-diol synthesis. J. Am. Chem. Soc. 2007, 129, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Momiyama, N.; Yamamoto, H. Bronsted acid catalysis of achiral enamine for regio- and enantioselective nitroso aldol synthesis. J. Am. Chem. Soc. 2005, 127, 1080–1081. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Candeias, N.R.; Barbas, C.F. Construction of bispirooxindoles containing three quaternary stereocentres in a cascade using a single multifunctional organocatalyst. Nat. Chem 2011, 3, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, C.; Sugiura, M.; Kobayashi, S. Stereospecific, enantioselective allylation of alpha-hydrazono esters by using allyltrichlorosilanes with BINAP dioxides as neutral-coordinate organocatalysts. Angew. Chem. Int. Ed. 2004, 43, 6491–6493. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Nakatsu, H.; Takiguchi, Y.; Maruoka, K. Axially Chiral Dicarboxylic Acid Catalyzed Activation of Quinone Imine Ketals: Enantioselective Arylation of Enecarbamates. J. Am. Chem. Soc. 2013, 135, 16010–16013. [Google Scholar] [CrossRef] [PubMed]
- McDougal, N.T.; Schaus, S.E. Asymmetric Morita-Baylis-Hillman reactions catalyzed by chiral Bronsted acids. J. Am. Chem. Soc. 2003, 125, 12094–12095. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, Y.; Gotanda, T.; Ito, K. Asymmetric Morita-Baylis-Hillman reactions of 2-cyclohexen-1-one catalyzed by chiral biaryl-based bis(thiourea) organocatalysts. Tetrahedron Lett. 2011, 52, 6234–6237. [Google Scholar] [CrossRef]
- Zhao, H.W.; Yue, Y.Y.; Li, H.L.; Song, X.Q.; Sheng, Z.H.; Yang, Z.; Meng, W.; Yang, Z. Novel Axially Unfixed Biaryl-Based Water-Compatible Organocatalysts: Design, Synthesis and Their Asymmetric Catalysis in Direct Aldol Reactions in Water. Synlett 2013, 24, 2160–2164. [Google Scholar] [CrossRef]
- Yeung, C.T.; Yeung, H.L.; Chan, W.T.K.; Yan, S.C.; Tam, E.C.Y.; Wong, K.L.; Lee, C.S.; Wong, W.T. Stereolabile chiral biphenyl hybrids: Crystallization-induced dynamic atropselective resolution involving supramolecular interactions. CrystEngComm 2013, 15, 836–840. [Google Scholar] [CrossRef]
- Omote, M.; Nishimura, Y.; Sato, K.; Ando, A.; Kumadaki, I. Synthesis of new axially dissymmetric ligand with large perfluoroalkyl groups. Tetrahedron Lett. 2005, 46, 319–322. [Google Scholar] [CrossRef]
- Omote, M.; Nishimura, Y.; Sato, K.; Ando, A.; Kumadaki, I. New axially dissymmetric ligand recoverable with fluorous solvent. Tetrahedron 2006, 62, 1886–1894. [Google Scholar] [CrossRef]
- Hasegawa, T.; Omote, M.; Sato, K.; Ando, A.; Kumadaki, I. New approach to a novel axially chiral ligand showing spontaneous enrichment of axial chirality. Chem. Pharm. Bull. 2003, 51, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Omote, M.; Sato, K.; Ando, A.; Kumadaki, I. Synthesis of axially dissymmetric ligands with two chiral centers of perfluoroalkyl carbinol moiety, and their application to asymmetric syntheses. Curr Org. Synth 2007, 4, 137–150. [Google Scholar] [CrossRef]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Atropisomers 2 are available upon reasonable request. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Catalysta | Solvent | Temperature | Enamine | Yield%b | Ee%c |
|---|---|---|---|---|---|---|
| 1 | 1 | Toluene | −50 °C | Morpholine enamine | 63 | 12 (S) |
| 2 | (S,Ra,S)-2 | Toluene | −50 °C | Morpholine enamine | 38 | 44 (S) |
| 3 | (S,Sa,S)-2 | Toluene | −50 °C | Morpholine enamine | 23 | 8 (S) |
| 4 | (S,Ra,S)-2 | Toluene | −50 °C | Piperidine enamine | 24 | 30 (S) |
| 5d | (S,Ra,S)-2 | Toluene | −50 °C | Pyrrolidine enamine | - | - |
| 6 | (S,Ra,S)-2 | DCM | −50 °C | Morpholine enamine | 55 | 14 (S) |
| 7 | (S,Ra,S)-2 | Et2O | −50 °C | Morpholine enamine | 62 | 41 (S) |
| 8 | (S,Ra,S)-2 | THF | −50 °C | Morpholine enamine | 64 | 31 (S) |
| 9 | (S,Ra,S)-2 | Toluene | −80 °C | Morpholine enamine | 64 | 50 (S) |
| 10e | (S,Ra,S)-2 | Toluene | −80 °C | Morpholine enamine | 58 | 46 (S) |
| 11f | (S,Ra,S)-2 | Toluene | −80 °C | Morpholine enamine | 47 | 34 (S) |
| 12 | (R,Sa,R)-2 | Toluene | −80 °C | Morpholine enamine | 61 | 48 (R) |
| 13 | (R,Ra,R)-2 | Toluene | −80 °C | Morpholine enamine | 53 | 20 (R) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeung, C.-T.; Chan, W.T.K.; Lo, W.-S.; Law, G.-L.; Wong, W.-T. Synthesis of a Conformationally Stable Atropisomeric Pair of Biphenyl Scaffold Containing Additional Stereogenic Centers. Molecules 2019, 24, 643. https://doi.org/10.3390/molecules24030643
Yeung C-T, Chan WTK, Lo W-S, Law G-L, Wong W-T. Synthesis of a Conformationally Stable Atropisomeric Pair of Biphenyl Scaffold Containing Additional Stereogenic Centers. Molecules. 2019; 24(3):643. https://doi.org/10.3390/molecules24030643
Chicago/Turabian StyleYeung, Chi-Tung, Wesley Ting Kwok Chan, Wai-Sum Lo, Ga-Lai Law, and Wing-Tak Wong. 2019. "Synthesis of a Conformationally Stable Atropisomeric Pair of Biphenyl Scaffold Containing Additional Stereogenic Centers" Molecules 24, no. 3: 643. https://doi.org/10.3390/molecules24030643