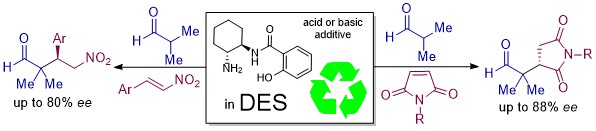
3.3. General Procedure for the Enantioselective Conjugate Addition of Aldehydes to Maleimides
To a mixture of catalyst
8 (4.7 mg, 0.02 mmol), 4-nitrobenzoic acid (3.3 mg, 0.02 mmol) and maleimide
10 (0.2 mmol) in ChCl/EG (1/2 molar ratio, 0.5 mL) was added the aldehyde
9 (0.4 mmol), and the reaction was vigorously stirred at rt until completion (TLC) (
Table 2). After this period, HCl 2N (10 mL) was added and the reaction product was extracted with AcOEt (3 × 10 mL). The combined organic phases were washed with saturated NaHCO
3 (10 mL) and brine (10 mL), dried over MgSO
4 and, after filtration, the solvent was evaporated under reduced pressure (15 torr) to get the crude product, which was purified by flash column chromatography on silica gel (
n-hexane/AcOEt gradients). The adducts 11 were identified by comparison of their NMR data with those of the literature (
Supplementary Materials, NMR spectra). Their enantiomeric excesses were determined by chiral HPLC on the reaction crude, using the conditions described in each case (
Supplementary Materials, HPLC chromatograms).
2-(2,5-Dioxo-1-phenylpyrrolidin-3-yl)-2-methylpropanal (
11aa) [
36]. White solid (48 mg, 98%);
1H NMR (CDCl
3): δ
H = 9.52 (s, 1H), 7.51−7.43 (m, 2H), 7.42–7.36 (m, 1H), 7.31−7.26 (m, 2H), 3.15 (dd,
J = 9.5, 5.5 Hz, 1H), 2.98 (dd,
J = 18.3, 9.5 Hz, 1H), 2.62 (dd,
J = 18.3, 5.5 Hz, 1H), 1.33 (s, 3H), 1.29 (s, 3H) ppm;
13C NMR (CDCl
3): δ
C = 202.9, 177.0, 174.9, 131.9, 129.3, 128.9, 126.7, 48.7, 45.1, 32.0, 20.5, 19.8 ppm; HPLC: Chiralcel OD-H, λ = 240 nm,
n-hexane/2-propanol, 80:20, 1.0 mL/min, t
r (
major) = 25.0 min, t
r (
minor) = 30.6 min.
2-(2,5-Dioxo-1-(p-tolyl)pyrrolidin-3-yl)-2-methylpropanal (
11ab) [
36]. White solid (46 mg, 88%);
1H NMR (CDCl
3): δ
H = 9.49 (s, 1H), 7.31−7.22 (m, 2H), 7.17−7.08 (m, 2H), 3.12 (dd,
J = 9.5, 5.5 Hz, 1H), 2.93 (dd,
J = 18.3, 9.5 Hz, 1H), 2.57 (dd,
J = 18.3, 5.5 Hz, 1H), 2.37 (s, 3H), 1.28 (s, 3H), 1.24 (s, 3H) ppm;
13C NMR (CDCl
3): δ
C = 202.9, 177.1, 175.0, 138.8, 129.9, 129.2, 126.4, 48.5, 45.0, 31.8, 21.2, 20.3, 19.4 ppm; HPLC: Chiralcel OD-H, λ = 230 nm,
n-hexane/2-propanol, 80:20, 1.0 mL/min, t
r (
major) = 19.4 min, t
r (
minor) = 22.6 min.
2-(1-(4-Methoxyphenyl)-2,5-dioxopyrrolidin-3-yl)-2-methylpropanal (
11ac) [
40]. White solid (54 mg, 98%);
1H NMR (CDCl
3): δ
H = 9.52 (s, 1H), 7.23−7.14 (m, 2H), 7.02−6.93 (m, 2H), 3.82 (s, 3H), 3.14 (dd,
J = 9.5, 5.4 Hz, 1H), 2.97 (dd,
J = 18.2, 9.5 Hz, 1H), 2.60 (dd,
J = 18.2, 5.4 Hz, 1H), 1.32 (s, 3H), 1.28 (s, 3H) ppm;
13C NMR (CDCl
3): δ
C = 202.9, 177.3, 175.2, 159.7, 127.9, 124.5, 114.6, 55.6, 48.6, 45.1, 31.9, 20.4, 19.7 ppm; HPLC: Chiralpak AS-H, λ = 240 nm,
n-hexane/2-propanol, 80:20, 1.0 mL/min, t
r (
major) = 31.0 min, t
r (
minor) = 34.8 min.
2-(1-(4-Chlorophenyl)-2,5-dioxopyrrolidin-3-yl)-2-methylpropanal (
11ad) [
36]. White solid (33 mg, 59%);
1H NMR (CDCl
3): δ
H = 9.49 (s, 1H), 7.51−7.38 (m, 2H), 7.31−7.20 (m, 2H), 3.11 (dd,
J = 9.5, 5.4 Hz, 1H), 2.97 (dd,
J = 18.1, 9.5 Hz, 1H), 2.61 (dd,
J = 18.1, 5.4 Hz, 1H), 1.36 (s, 3H), 1.29 (s, 3H) ppm;
13C NMR (CDCl
3): δ
C = 202.8, 176.8, 174.6, 134.6, 130.4, 129.5, 127.9, 48.8, 45.1, 32.1, 20.6, 20.0 ppm; HPLC: Chiralcel OD-H, λ = 230 nm,
n-hexane/2-propanol, 80:20, 1.0 mL/min, t
r (
major) = 21.2 min, t
r (
minor) = 35.6 min.
2-(1-(4-Bromophenyl)-2,5-dioxopyrrolidin-3-yl)-2-methylpropanal (
11ae) [
36]. White solid (55 mg, 85%);
1H NMR (CDCl
3): δ
H = 9.48 (s, 1H), 7.64−7.55 (m, 2H), 7.23−7.14 (m, 2H), 3.11 (dd,
J = 9.5, 5.5 Hz, 1H), 2.97 (dd,
J = 18.2, 9.5 Hz, 1H), 2.61 (dd,
J = 18.2, 5.5 Hz, 1H), 1.35 (s, 3H), 1.28 (s, 3H) ppm;
13C NMR (CDCl
3): δ
C = 202.8, 176.7, 174.5, 132.5, 130.9, 128.2, 122.7, 48.8, 45.1, 32.1, 20.6, 20.0 ppm; HPLC: Chiralcel OD-H, λ = 240 nm,
n-hexane/2-propanol, 80:20, 1.0 mL/min, t
r (
major) = 22.2 min, t
r (
minor) = 34.9 min.
2-Methyl-2-(1-methyl-2,5-dioxopyrrolidin-3-yl)propanal (
11af) [
22]. White solid (36 mg, 98%);
1H NMR (CDCl
3): δ
H = 9.51 (s, 1H), 3.05 (dd,
J = 9.2, 5.2 Hz, 1H), 2.99 (s, 3H), 2.83 (dd,
J = 18.2, 9.2 Hz, 1H), 2.45 (dd,
J = 18.2, 5.2 Hz, 1H), 1.22 (s, 3H), 1.21 (s, 3H) ppm;
13C NMR (CDCl
3): δ
C = 202.9, 177.9, 175.9, 48.0, 45.1, 31.5, 24.9, 20.1, 19.2 ppm; HPLC: Chiralpak AS-H, λ = 210 nm,
n-hexane/2-propanol, 80:20, 1.0 mL/min, t
r (
minor) = 11.5 min, t
r (
major) = 12.5 min.
2-(2,5-Dioxopyrrolidin-3-yl)-2-methylpropanal (
11ag) [
22]. White solid (33 mg, 98%);
1H NMR (CDCl
3): δ
H = 9.49 (s, 1H), 9.03 (br. s, 1H), 3.10 (dd,
J = 7.7, 5.5 Hz, 1H), 2.85 (dd,
J = 18.3, 7.7 Hz, 1H), 2.50 (dd,
J = 18.3, 5.5 Hz, 1H), 1.24 (s, 3H), 1.23 (s, 3H) ppm;
13C NMR (CDCl
3): δ
C = 203.0, 178.5, 176.4, 48.1, 46.4, 32.9, 20.2, 19.4 ppm; HPLC: Chiralpak AD-H, λ = 210 nm,
n-hexane/2-propanol, 80:20, 1.0 mL/min, t
r (
minor) = 16.8 min, t
r (
major) = 21.2 min.
1-(2,5-Dioxo-1-phenylpyrrolidin-3-yl)cyclohexane-1-carbaldehyde (
11ba) [
36]. White solid (36 mg, 63%);
1H NMR (CDCl
3): δ
H = 9.54 (s, 1H), 7.56−7.33 (m, 3H), 7.33−7.26 (m, 2H), 3.22 (dd,
J = 9.4, 6.0 Hz, 1H), 2.88 (dd,
J = 18.1, 9.4 Hz, 1H), 2.68 (dd,
J = 18.1, 6.0 Hz, 1H), 1.96 (m, 2H), 1.60 (m, 8H) ppm;
13C NMR (CDCl
3): δ
C = 204.7, 177.2, 174.9, 132.1, 129.3, 128.8, 126.8, 52.3, 42.9, 31.7, 28.8, 28.3, 25.3, 21.6, 21.4 ppm; HPLC: Chiralcel OD-H, λ = 240 nm,
n-hexane/2-propanol, 80:20, 1.0 mL/min, t
r (
major) = 23.9 min, t
r (
minor) = 30.9 min.
3.4. General Procedure for the Enantioselective Conjugate Addition of Isobutyraldehyde to Nitroalkenes
To a mixture of catalyst
8 (4.7 mg, 0.02 mmol), DMAP (2.4 mg, 0.02 mmol) and nitroalkene
12 (0.2 mmol) in ChCl/H
2O (1/2 molar ratio, 0.5 mL) was added isobutyraldehyde
9a (37 µL, 28.8 mg, 0.4 mmol), and the reaction was vigorously stirred at rt until completion (TLC) (
Table 4). After this period, HCl 2N (10 mL) was added and the reaction product was extracted with AcOEt (3 × 10 mL). The combined organic phases were washed with saturated NaHCO
3 (10 mL) and brine (10 mL), dried over MgSO
4 and, after filtration, the solvent was evaporated under reduced pressure (15 torr) to get the crude product, which was purified by flash column chromatography on silica gel (
n-hexane/AcOEt gradients). The adducts 13a were identified by comparison of their NMR data with those of the literature (
Supplementary Materials, NMR spectra). Their enantiomeric excesses were determined by chiral HPLC on the reaction crude, using the conditions described in each case (
Supplementary Materials, HPLC chromatograms).
2,2-Dimethyl-4-nitro-3-phenylbutanal (
13aa) [
37]. Yellow oil (41 mg, 92%);
1H NMR (CDCl
3): δ
H = 9.53 (s, 1H), 7.39−7.28 (m, 3H), 7.24−7.15 (m, 2H), 4.86 (dd,
J = 13.0, 11.2 Hz, 1H), 4.69 (dd,
J = 13.0, 4.3 Hz, 1H), 3.78 (dd,
J = 11.2, 4.3 Hz, 1H), 1.14 (s, 3H), 1.01 (s, 3H) ppm;
13C NMR (CDCl
3): δ
C = 204.4, 135.5, 129.2, 128.9, 128.3, 76.5, 48.7, 48.4, 21.8, 19.1 ppm; HPLC: Chiralcel OD-H, λ = 210 nm,
n-hexane/2-propanol, 80:20, 1.0 mL/min, t
r (
major) = 12.2 min, t
r (
minor) = 17.4 min.
2,2-Dimethyl-4-nitro-3-(p-tolyl)butanal (
13ab) [
41]. Yellow oil (28 mg, 60%);
1H NMR (CDCl
3): δ
H = 9.53 (s, 1H), 7.18–7.03 (m, 4H), 4.83 (dd,
J = 12.9, 11.3 Hz, 1H), 4.67 (dd,
J = 12.9, 4.3 Hz, 1H), 3.74 (dd,
J = 11.3, 4.3 Hz, 1H), 2.32 (s, 3H), 1.13 (s, 3H), 1.00 (s, 3H) ppm;
13C NMR (CDCl
3): δ
C = 204.5, 138.1, 132.4, 129.6, 129.1, 76.6, 48.5, 48.4, 21.8, 21.2, 19.1 ppm; HPLC: Chiralcel OD-H, λ = 240 nm,
n-hexane/2-propanol, 80:20, 1.0 mL/min, t
r (
major) = 9.2 min, t
r (
minor) = 12.4 min.
3-(4-Methoxyphenyl)-2,2-dimethyl-4-nitrobutanal (
13ac) [
37]. Yellow oil (37 mg, 74%);
1H NMR (CDCl
3): δ
H = 9.52 (s, 1H), 7.17−7.05 (m, 2H), 6.91−6.79 (m, 2H), 4.81 (dd,
J = 12.8, 11.3 Hz, 1H), 4.66 (dd,
J = 12.8, 4.3 Hz, 1H), 3.79 (s, 3H), 3.73 (dd,
J = 11.3, 4.3 Hz, 1H), 1.12 (s, 3H), 1.00 (s, 3H) ppm;
13C NMR (CDCl
3): δ
C = 204.5, 159.5, 130.3, 127.3, 114.3, 76.7, 55.4, 48.5, 48.1, 21.7, 19.1 ppm; HPLC: Chiralcel OD-H, λ = 240 nm,
n-hexane/2-propanol, 80:20, 1.0 mL/min, t
r (
major) = 12.1 min, t
r (
minor) = 16.4 min.
3-(Benzo[d] [
1,
3]
dioxol-5-yl)-2,2-dimethyl-4-nitrobutanal (
13ad) [
25]. Yellow oil (36 mg, 67%);
1H NMR (CDCl
3): δ
H = 9.51 (s, 1H), 6.75 (d,
J = 7.9 Hz, 1H), 6.72−6.61 (m, 2H), 5.96 (s, 2H), 4.78 (dd,
J = 12.9, 11.3 Hz, 1H), 4.65 (dd,
J = 12.9, 4.3 Hz, 1H), 3.69 (dd,
J = 11.3, 4.3 Hz, 1H), 1.13 (s, 3H), 1.02 (s, 3H) ppm;
13C NMR (CDCl
3): δ
C = 204.4, 148.1, 147.6, 129.1, 122.8, 109.3, 108.5, 101.4, 76.7, 48.5 (2xC), 21.8, 19.2 ppm; HPLC: Chiralcel OD-H, λ = 230 nm,
n-hexane/2-propanol, 80:20, 1.0 mL/min, t
r (
major) = 15.8 min, t
r (
minor) = 20.5 min.
2,2-Dimethyl-4-nitro-3-(3,4,5-trimethoxyphenyl)butanal (
13ae) [
25]. Yellow oil (45 mg, 73%);
1H NMR (CDCl
3): δ
H = 9.52 (s, 1H), 6.38 (s, 2H), 4.85 (dd,
J = 13.0, 11.2 Hz, 1H), 4.69 (dd,
J = 13.0, 4.3 Hz, 1H), 3.84 (s, 6H), 3.83 (s, 3H), 3.69 (dd,
J = 11.2, 4.3 Hz, 1H), 1.16 (s, 3H), 1.06 (s, 3H) ppm;
13C NMR (CDCl
3): δ
C = 204.5, 153.6, 138.1, 131.2, 106.6, 76.6, 61.0, 56.4, 49.2, 48.4, 22.0, 19.6 ppm; HPLC: Chiralcel OD-H, λ = 210 nm,
n-hexane/2-propanol, 80:20, 1.0 mL/min, t
r (
major) = 15.5 min, t
r (
minor) = 17.8 min.
3-(4-Fluorophenyl)-2,2-dimethyl-4-nitrobutanal (
13af) [
37]. Yellow oil (76 mg, 78%);
1H NMR (CDCl
3): δ
H = 9.51 (s, 1H), 7.24−7.13 (m, 2H), 7.09−6.98 (m, 2H), 4.82 (dd,
J = 13.0, 11.3 Hz, 1H), 4.69 (dd,
J = 13.0, 4.3 Hz, 1H), 3.78 (dd,
J = 11.3, 4.3 Hz, 1H), 1.13 (s, 3H), 1.01 (s, 3H) ppm;
13C NMR (CDCl
3): δ
C = 204.1, 163.6, 161.6, 130.9, 130.8, 116.0, 115.8, 76.5, 48.4, 48.1, 21.8, 19.1 ppm; HPLC: Chiralcel OD-H, λ = 210 nm,
n-hexane/2-propanol, 80:20, 1.0 mL/min, t
r (
major) = 10.0 min, t
r (
minor) = 15.6 min.
3-(2-Chlorophenyl)-2,2-dimethyl-4-nitrobutanal (
13ag) [
42]. Yellow oil (25 mg, 49%);
1H NMR (CDCl
3): δ
H = 9.55 (s, 1H), 7.45–7.39 (m, 1H), 7.31–7.26 (m, 2H), 7.23 (m, 1H), 4.97–4.48 (m, 3H), 1.17 (s, 3H), 1.07 (s, 3H) ppm;
13C-NMR (CDCl
3): δ
C = 203.9, 135.9, 133.9, 130.6, 129.3, 128.4, 127.3, 76.3, 49.2, 42.6, 21.0, 18.8 ppm; HPLC: Chiralcel OD-H, λ = 210 nm,
n-hexane/2-propanol, 80:20, 1.0 mL/min, t
r (
major) = 9.7 min, t
r (
minor) = 23.2 min.
3-(4-Chlorophenyl)-2,2-dimethyl-4-nitrobutanal (
13ah) [
37]. Yellow oil (45 mg, 89%);
1H NMR (CDCl
3): δ
H = 9.50 (s, 1H), 7.35−7.29 (m, 2H), 7.18−7.11 (m, 2H), 4.83 (dd,
J = 13.1, 11.3 Hz, 1H), 4.69 (dd,
J = 13.1, 4.2 Hz, 1H), 3.77 (dd,
J = 11.3, 4.2 Hz, 1H), 1.13 (s, 3H), 1.01 (s, 3H) ppm;
13C-NMR (CDCl
3): δ
C = 204.0, 134.3, 134.1, 130.5, 129.1, 76.3, 48.3, 48.1, 21.9, 19.1 ppm; HPLC: Chiralcel OD-H, λ = 210 nm,
n-hexane/2-propanol, 80:20, 1.0 mL/min, t
r (
major) = 11.2 min, t
r (
minor) = 16.4 min.
3-(2-Bromophenyl)-2,2-dimethyl-4-nitrobutanal (
13ai) [
41]. Yellow oil (42 mg, 70%);
1H NMR (CDCl
3): δ
H = 9.55 (s, 1H), 7.61 (dd,
J = 8.0, 1.2 Hz, 1H), 7.36–7.25 (m, 2H), 7.15 (ddd,
J = 8.0, 6.8, 2.2 Hz, 1H), 4.84 (dd,
J = 13.2, 11.3 Hz, 1H), 4.72 (dd,
J = 13.2, 4.1 Hz, 1H), 4.62 (dd,
J = 11.3, 4.1 Hz, 1H), 1.17 (s, 3H), 1.09 (s, 3H) ppm;
13C-NMR (CDCl
3): δ
C = 203.9, 135.7, 134.0, 129.6, 128.5, 128.0, 127.2, 76.6, 49.2, 45.4, 21.1, 18.9 ppm; HPLC: Chiralcel OD-H, λ = 230 nm,
n-hexane/2-propanol, 80:20, 1.0 mL/min, t
r (
major) = 10.5 min, t
r (
minor) = 25.6 min.
3-(4-Bromophenyl)-2,2-dimethyl-4-nitrobutanal (
13aj) [
37]. Yellow oil (50 mg, 83%);
1H NMR (CDCl
3): δ
H = 9.50 (s, 1H), 7.51−7.42 (m, 2H), 7.15−7.04 (m, 2H), 4.82 (dd,
J = 13.1, 11.3 Hz, 1H), 4.69 (dd,
J = 13.1, 4.2 Hz, 1H), 3.76 (dd,
J = 11.3, 4.2 Hz, 1H), 1.12 (s, 3H), 1.01 (s, 3H) ppm;
13C NMR (CDCl
3): δ
C = 203.9, 134.7, 132.0, 130.9, 122.4, 76.2, 48.2, 48.1, 21.9, 19.1 ppm; HPLC: Chiralcel OD-H, λ = 230 nm,
n-hexane/2-propanol, 80:20, 1.0 mL/min, t
r (
major) = 12.4 min, t
r (
minor) = 17.4 min.
2,2-Dimethyl-4-nitro-3-(4-(trifluoromethyl)phenyl)butanal (
13ak) [
43]. Yellow oil (49 mg, 85%);
1H NMR (CDCl
3): δ
H = 9.50 (s, 1H), 7.61 (d,
J = 8.1 Hz, 2H), 7.36 (d,
J = 8.1 Hz, 2H), 4.89 (dd,
J = 13.3, 11.4 Hz, 1H), 4.74 (dd,
J = 13.3, 4.1 Hz, 1H), 3.87 (dd,
J = 11.4, 4.1 Hz, 1H), 1.15 (s, 3H), 1.03 (s, 3H) ppm;
13C NMR (CDCl
3): δ
C = 203.7, 139.9, 129.7, 125.9 (2xC), 125.8 (2xC), 76.1, 48.4, 48.3, 22.0, 19.1 ppm; HPLC: Chiralcel OD-H, λ = 210 nm,
n-hexane/2-propanol, 80:20, 1.0 mL/min, t
r (
major) = 10.4 min, t
r (
minor) = 16.2 min.
2,2-Dimethyl-3-(naphthalen-2-yl)-4-nitrobutanal (
13al) [
37]. Yellow oil (36 mg, 67%);
1H NMR (CDCl
3): δ
H = 9.55 (s, 1H), 7.84−7.77 (m, 3H), 7.66 (d,
J = 1.4 Hz, 1H), 7.52−7.45 (m, 2H), 7.31 (dd,
J = 8.6, 1.9 Hz, 1H), 4.98 (dd,
J = 13.1, 11.3 Hz, 1H), 4.76 (dd,
J = 13.1, 4.2 Hz, 1H), 3.95 (dd,
J = 11.3, 4.2 Hz, 1H), 1.17 (s, 3H), 1.04 (s, 3H) ppm;
13C NMR (CDCl
3): δ
C = 204.4, 133.2, 133.1, 133.0, 128.6, 128.5, 128.0, 127.8, 126.7, 126.6, 126.5, 76.5, 48.8, 48.6, 22.0, 19.2 ppm; HPLC: Chiralcel OD-H, λ = 280 nm,
n-hexane/2-propanol, 75:25, 1.0 mL/min, t
r (
major) = 15.6 min, t
r (
minor) = 18.1 min.
3-(Furan-2-yl)-2,2-dimethyl-4-nitrobutanal (
13am) [
37]. Yellow oil (32 mg, 75%);
1H NMR (CDCl
3): δ
H = 9.52 (s, 1H), 7.37 (d,
J = 1.9 Hz, 1H), 6.32 (dd,
J = 3.2, 1.9 Hz, 1H), 6.22 (dd,
J = 7.1, 3.2 Hz, 1H), 4.76 (dd,
J = 12.9, 11.0 Hz, 1H), 4.59 (dd,
J = 12.9, 3.9 Hz, 1H), 3.92 (dd,
J = 11.0, 3.9 Hz, 1H), 1.18 (s, 3H), 1.05 (s, 3H) ppm;
13C NMR (CDCl
3): δ
C = 203.6, 149.9, 142.9, 110.6, 109.8, 75.0, 48.3, 42.4, 21.3, 19.3 ppm; HPLC: Chiralcel OD-H, λ = 230 nm,
n-hexane/2-propanol, 80:20, 1.0 mL/min, t
r (
major) = 8.1 min, t
r (
minor) = 11.8 min.








{kind=link}
{kind=link}
{kind=link}



