
Oxovanadium(IV) Coordination Compounds with Kojic Acid Derivatives in Aqueous Solution

 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Potentiometric Data
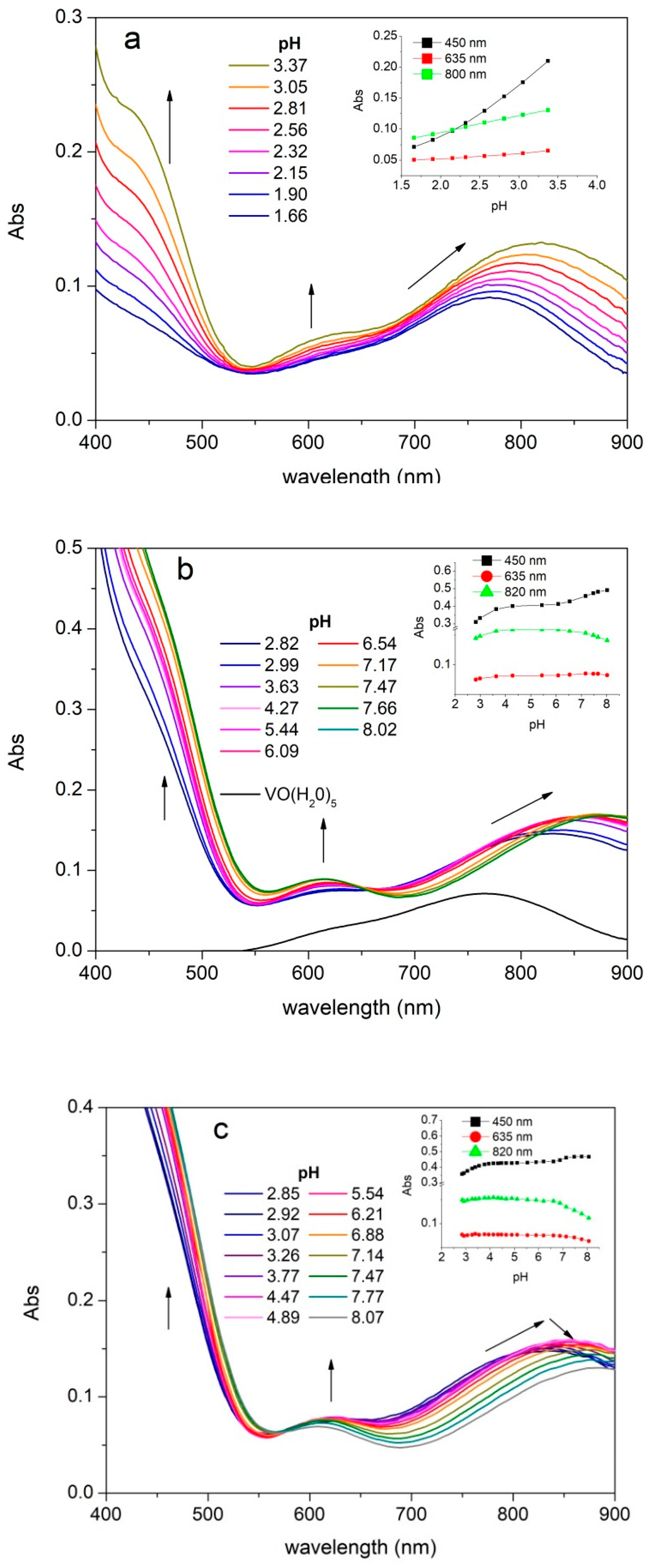
2.2. UV-Visible Spectra
2.3. EPR Spectra
3. Sequestering Capability
4. Materials and Methods
4.1. Chemicals
4.2. Potentiometric Measurements
4.3. Spectroscopic Measurements
4.4. Data Analysis and Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Crans, D.C.; Smee, J.J.; Gaidamauskas, E.; Yang, L. The chemistry and biochemistry of vanadium and the biological activities exerted by vanadium compounds. Chem. Rev. 2004, 104, 849–902. [Google Scholar] [CrossRef] [PubMed]
- Sanna, D.; Bíró, L.; Buglyó, P.; Micera, G.; Garribba, E. Transport of the anti-diabetic VO2+ complexes formed by pyrone derivatives in the blood serum. J. Inorg. Biochem. 2012, 115, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.H.; Orvig, C. Coordination chemistry of vanadium in metallopharmaceutical candidate compounds. Coord. Chem. Rev. 2001, 219–221, 1033–1053. [Google Scholar] [CrossRef]
- Zoroddu, M.A.; Aaseth, J.; Crisponi, G.; Medici, S.; Peana, M.; Nurchi, V.M. The essential metals for humans: A brief overview. J. Inorg. Biochem. 2019, 195, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.H.; Lichter, J.; LeBel, C.; Scaife, M.C.; McNeill, J.H.; Orvig, C. Vanadium treatment of type 2 diabetes: A view to the future. J. Inorg. Biochem. 2009, 103, 554–558. [Google Scholar] [CrossRef]
- Crans, D.C.; LaRee, H.; Cardiff, G.; Posner, B. Essential metals in medicine: Therapeutic use and toxicity of metal ions in the clinic. In Essential Metals in Medicine: Therapeutic Use and Toxicity of Metal; Astrid Siegel, Ed.; De Gruyter: Berlin, Germany, 2019; pp. 203–230. ISBN 3110527871. [Google Scholar]
- Yuen, V.G.; Caravan, P.; Gelmini, L.; Glover, N.; McNeill, J.H.; Setyawati, I.A.; Zhou, Y.; Orvig, C.; Chaudhary, J.; Pathak, A.N.; et al. Pyrone derivatives and metals: From natural products to metal-based drugs. J. Inorg. Biochem. 1997, 104, 94–106. [Google Scholar]
- Kandioller, W.; Kurzwernhart, A.; Hanif, M.; Meier, S.M.; Henke, H.; Keppler, B.K.; Hartinger, C.G. Pyrone derivatives and metals: From natural products to metal-based drugs. J. Organomet. Chem. 2011, 696, 999–1010. [Google Scholar] [CrossRef]
- Yuen, V.G.; Caravan, P.; Gelmini, L.; Glover, N.; McNeill, J.H.; Setyawati, I.A.; Zhou, Y.; Orvig, C. Glucose-lowering properties of vanadium compounds: Comparison of coordination complexes with maltol or kojic acid as ligands. J. Inorg. Biochem. 1997, 68, 109–116. [Google Scholar] [CrossRef]
- Wei, Y.; Zhang, C.; Zhao, P.; Yang, X.; Wang, K. A new salicylic acid-derivatized kojic acid vanadyl complex: Synthesis, characterization and anti-diabetic therapeutic potential. J. Inorg. Biochem. 2011, 105, 1081–1085. [Google Scholar] [CrossRef]
- Wei, Y.B.; Yang, X. Da Synthesis, characterization and anti-diabetic therapeutic potential of a new benzyl acid-derivatized kojic acid vanadyl complex. BioMetals 2012, 25, 1261–1268. [Google Scholar] [CrossRef]
- Nurchi, V.M.; Crisponi, G.; Lachowicz, J.I.; de Guadalupe Jaraquemada-Pelaez, M.; Bretti, C.; Peana, M.; Medici, S.; Zoroddu, M.A. Equilibrium studies of new bis-hydroxypyrone derivatives with Fe3+, Al3+, Cu2+ and Zn2+. J. Inorg. Biochem. 2018, 189, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Nurchi, V.M.; de Guadalupe Jaraquemada-Pelaez, M.; Crisponi, G.; Lachowicz, J.I.; Cappai, R.; Gano, L.; Santos, M.A.; Melchior, A.; Tolazzi, M.; Peana, M.; et al. A new tripodal kojic acid derivative for iron sequestration: Synthesis, and in vivo bioassays. J. Inorg. Biochem. 2019, 193, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- De Juan, A.; Jaumot, J.; Tauler, R. Multivariate curve resolution (MCR). Solving the mixture analysis problem. Anal. Methods 2014, 6, 4964–4976. [Google Scholar] [CrossRef]
- Komura, A.; Hayashi, M.; Hiroto, I. Hydrolytic behavior of oxovanadium(IV) ions. Bull. Chem. Soc. Jpn. 1977, 50, 2927–2931. [Google Scholar] [CrossRef]
- De Stefano, C.; Mineo, P.; Rigano, C.; Sammartano, S. Ionic strength dependence of formation constants. XVII: The calculation of equilibrium concentrations and formation. Ann. Chim. 1993, 83, 243–277. [Google Scholar]
- Buglyó, P.; Kiss, E.; Fábián, I.; Kiss, T.; Sanna, D.; Garribba, E.; Micera, G. Speciation and NMR relaxation studies of VO(IV) complexes with several O-donor containing ligands: Oxalate, malonate, maltolate and kojate. Inorganica Chim. Acta 2000, 306, 174–183. [Google Scholar] [CrossRef]
- Song, B.; Saatchi, K.; Rawji, G.H.; Orvig, C. Synthesis and solution studies of the complexes of pyrone analogue ligands with vanadium(IV) and vanadium(V). Inorganica Chim. Acta 2002, 339, 393–399. [Google Scholar] [CrossRef]
- Chruscinska, E.; Garribba, E.; Micera, G.; Panzanelli, A. L-mimosine, an amino acid with maltol-type binding properties toward copper(II), oxovanadium(IV) and other metal ions. J. Inorg. Biochem. 1999, 75, 225–232. [Google Scholar] [CrossRef]
- Berto, S.; Daniele, P.G.; Foti, C.; Prenesti, E.; Sammartano, S. Interaction of oxovanadium(IV) with carboxylic ligands in aqueous solution: A thermodynamic and visible spectrophotometric study. J. Mol. Liq. 2008, 142, 57–63. [Google Scholar] [CrossRef]
- Mabbs, F.E.; Collins, D. The use of matrix diagonalization in the simulation of the EPR powder spectra of D-transition metal compounds. Mol. Phys. Rep. 1999, 26, 39–59. [Google Scholar] [CrossRef]
- Spałek, T.; Pietrzyk, P.; Sojka, Z. Application of the genetic algorithm joint with the powell method to nonlinear least-squares fitting of powder EPR spectra. 2004. [Google Scholar] [CrossRef]
- Abou Fadel, M.; de Juan, A.; Touati, N.; Vezin, H.; Duponchel, L. New chemometric approach MCR-ALS to unmix EPR spectroscopic data from complex mixtures. J. Magn. Reson. 2014, 248, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Bonomo, R.P.; Riggi, F.; Di Bilio, A.J. EPR reinvestigation of the copper(II)-imidazole system. Inorg. Chem. 1988, 27, 2510–2512. [Google Scholar] [CrossRef]
- Smith, T.S.; Lobrutto, R.; Pecoraro, V.L. Paramagnetic spectroscopy of vanadyl complexes and its applications to biological systems. Coord. Chem. Rev. 2002, 228, 1–18. [Google Scholar] [CrossRef]
- Dessì, A.; Micera, G.; Sanna, D. EPR investigation of the oxovanadium(IV) complexes formed by the tripeptide glutathione and some related ligands in aqueous solution. J. Inorg. Biochem. 1993, 52, 275–286. [Google Scholar] [CrossRef]
- Yen, T.F.; Bucher, L.J. Electron Spin Resonance of Metal Complexes: Proceedings of the Symposium on ESR of Metal Chelates at the Pittsburgh Conference on Analytical Chemistry and Applied Spectroscopy, Held in Cleveland, Ohio, March 4–8, 1968; Springer US: New York, NY, USA, 1969; ISBN 1468483234. [Google Scholar]
- Crea, F.; De Stefano, C.; Foti, C.; Milea, D.; Sammartano, S. Chelating agents for the sequestration of mercury(II) and monomethyl mercury(II). Curr. Med. Chem. 2014, 21, 3819–3836. [Google Scholar] [CrossRef]
- Casale, A.; Daniele, P.G.; De Robertis, A.; Sammartano, S. Ionic strength dependence of formation constants. Part XI. An analysis of literature data on carboxylate ligand complexes. Ann. Chim. Chim. 1988, 78, 249. [Google Scholar]
- De Stefano, C.; Princi, P.; Rigano, C.; Sammartano, S. Computer-analysis of equilibrium data in solution Esab2M—An improved version of the Esab program. Ann. Chim. 1987, 77, 643–675. [Google Scholar]
- Pilbrow, J.R.; Winfield, M.E. Computer simulation of low symmetry ESR spectra due to vitamin B 12r and model systems. Mol. Phys. 1973, 25, 1073–1092. [Google Scholar] [CrossRef]
- Tauler, R. Multivariate curve resolution applied to second order data. Chemom. Intell. Lab. Syst. 1995, 30, 133–146. [Google Scholar] [CrossRef]
- Elbergali, A.; Nygren, J.; Kubista, M. An automated procedure to predict the number of components in spectroscopic data. Anal. Chim. Acta 1999, 379, 143–158. [Google Scholar] [CrossRef]
- De Juan, A.; Tauler, R. Chemometrics applied to unravel multicomponent processes and mixtures: Revisiting latest trends in multivariate resolution. Anal. Chim. Acta 2003, 500, 195–210. [Google Scholar] [CrossRef]
- Jaumot, J.; Gargallo, R.; de Juan, A.; Tauler, R. A graphical user-friendly interface for MCR-ALS: A new tool for multivariate curve resolution in MATLAB. Chemom. Intell. Lab. Syst. 2005, 76, 101–110. [Google Scholar] [CrossRef]
- Ruckebusch, C.; Blanchet, L. Multivariate curve resolution: A review of advanced and tailored applications and challenges. Anal. Chim. Acta 2013, 765, 28–36. [Google Scholar] [CrossRef]
- Jaumot, J.; de Juan, A.; Tauler, R. MCR-ALS GUI 2.0: New features and applications. Chemom. Intell. Lab. Syst. 2015, 140, 1–12. [Google Scholar] [CrossRef]
- Windig, W.; Guilment, J. Interactive self-modeling mixture analysis. Anal. Chem. 1991, 63, 1425–1432. [Google Scholar] [CrossRef]
- Ruckebusch, C. Resolving Spectral Mixtures: With Applications from Ultrafast Time-Resolved Spectroscopy to Super-Resolution Imaging; Elsevier: Amsterdam, The Netherlands, 2016; ISBN 9780444636447. [Google Scholar]
- Bro, R.; De Jong, S. A fast non-negativity-constrained least squares algorithm. J. Chemom. 1997, 11, 393–401. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds S2, S3, S4 and SC are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | S2 a | S3 a | S4 a | SC a | |
|---|---|---|---|---|---|
| logK for the reaction H+ + LHrz ⇆ Hr+1Lz+1 b | |||||
| Species | Species | ||||
| [LH]− | 9.85 | 10.20 | 10.59 | [LH]2− | 10.18 |
| [LH2]0 | 8.10 * | 8.87 | 9.56 | [LH2]− | 9.65 |
| [LH3]+ | 7.56 * | 7.94 * | 8.02 * | [LH3]0 | 8.72 |
| [LH4]2+ | 6.89 | 7.27 * | 7.38 * | [LH4]+ | 8.04* |
| [LH5]2+ | 7.70* | ||||
| [LH6]3+ | 7.10* | ||||
| logβ ± std dev c for reaction pVO2+ + qLz− + rH+ ⇆ (VO)pLqHr2p+r−qz | |||||
| Species | Species | ||||
| [VOLH3]3+ | 31.64 ± 0.01 | 35.03 ± 0.08 | 36.6 ± 0.1 | [VOLH5]4+ | 51.5 ± 0.08 |
| [VOLH2]2+ | 24.57 ± 0.02 | 31.89 ± 0.08 | 33.20 ± 0.08 | [VOLH4]3+ | 48.28 ± 0.03 |
| [VOLH]+ | - | 22.7 ± 0.1 | 23.5 ± 0.1 | [VOLH3]2+ | 40.8 ± 0.08 |
| logK for the reaction VO2+ + HrLz ⇆ (VO)LHr2−z | |||||
| Species | Species | ||||
| [VOLH3]3+ | 6.13 | 8.02 | 8.43 | [VOLH5]4+ | 7.21 |
| [VOLH2]2+ | 6.62 | 12.82 | 13.05 | [VOLH4]3+ | 11.69 |
| [VOLH]+ | - | 12.50 | 12.91 | [VOLH3]2+ | 12.25 |
| logK | Experimental Conditions | Reference | |||
| VO2+ + ka− ⇆ VOka+ | 7.63 | I = 0.2 mol L−1 KCl T = 25 °C | [18] | ||
| 7.61 | [9] | ||||
| VOka+ + ka− ⇆ VO(ka)2 | 6.74 | I = 0.2 mol L−1 KCl T = 25 °C | [18] | ||
| 6.89 | [9] | ||||
| Species a | Donor Set | Absorption Band | References | |||||
|---|---|---|---|---|---|---|---|---|
| Band I | Band II | Band III b | ||||||
| λmax | εmax | λmax | εmax | λmax | εmax | |||
| [VO(H2O)5]2+ | 765 | 16 | 635 | 7 | [21] | |||
| VO(ka)2 | 2 × (CO, O−) | 850 | 28 | 625 | 13 | 425 | 65 | [18] |
| [VO(ka)2OH]− | 2 × (CO, O−), OH− | 870 | 27 | 595 | 20 | 450sh | ||
| [VO(malt)]+ | (CO, O−) | 815 | 21 | 615 | 9 | 440 | 36 | [20] |
| VO(malt)2 | 2 × (CO, O−) | 860 | 27 | 620 | 12 | 440 | 73 | |
| [VO(malt)2(OH)]− | 2 × (CO, O−), OH− | 870 | 20 | 605 | 12 | 450 | 76 | |
| VO(HL-Mim)2 | 2 × (CO, O−) | 860 | 24 | 620 | 16 | 540 | 11 | |
| [VO(L-Mim)2]2− | 2 × (CO, O−) | 880 | 18 | 615 | 15 | 535 | 11 | |
| [VO(S2)H3]3+ | (CO, O−) | 851 | 34.3 ± 0.1 | ~612 sh c | ~440 | This work | ||
| [VO(S2)H2]2+ | (CO, O−), OH− | 886 | 35.5 ± 0.3 | 612 | 19.1 ± 0.3 | ~455 sh | ||
| [VO(S3)H3]3+ | (CO, O−) | 817 | 28.7 ± 0.4 | ~613 sh | ~452 sh | This work | ||
| [VO(S3)H2]2+ | 2 × (CO, O−) | 860 | 33.8 ± 0.2 | 622 | 16.8 ± 0.3 | ~455 sh | ||
| [VO(S3)H] | 2 × (CO, O−), OH− | 897 | 35 ± 1 | 592 | 21 ± 2 | ~469 | ||
| [VO(S4)H3]3+ | (CO, O−) | 819 | 28.6 ± 0.5 | 630 | 15.9 ± 0.6 | - | This work | |
| [VO(S4)H2]2+ | 2 × (CO, O−) | 861 | 33.1 ± 0.2 | 619 | 17.5 ± 0.3 | ~455 sh | ||
| [VO(S4)H] | 2 × (CO, O−), OH− | 898 | 29 ± 2 | ~615 sh | ~480 sh | |||
| [VO(SC)H5]4+ | (CO, O−) | 846 | 35.2 ± 09 | 612 sh | ~455 sh | This work | ||
| [VO(SC)H4]3+ | 2 × (CO, O−) | 854 | 32.0 ± 0.4 | 619 | 16.7 ± 0.4 | ~455 sh | ||
| [VO(SC)H3]2+ | 2 × (CO, O−), OH− | 884 | 34.8 ± 0.5 | 606 sh | ~480 sh | |||
| Ligand a | pH | SIM32 | MCR–ALS | ||||
|---|---|---|---|---|---|---|---|
| g0 | A0 (cm−1 ∙ 10−4) | R b | g0 | A0 (cm−1 ∙ 10−4) | R b | ||
| H2O | - | 1.965 | 106.9 | 0.993 | 1.970 | 105.9 | 0.990 |
| VO(H2O)5; VOLHn and n−1 | VO(H2O)5; VOLHn; VOLHn−1 | ||||||
| S2 | 2.5 | 1.964; 1968 | 104.6; 96.9 | 0.989 | 1.970; 1.975; 1.966 | 105.9; 97.7; 90.5 | 0.986 |
| 3.3 | 1.964; 1.967 | 104.3; 96.0 | 0.985 | 0.984 | |||
| 6.5 | 1.967 | 95.8 | 0.989 | 0.992 | |||
| 7.6 | 1.967 | 94.4 | 0.985 | 0.989 | |||
| 8.1 | 1.967 | 93.7 | 0.983 | 0.991 | |||
| S3 | 2.5 | 1.965; 1.968 | 104.8; 96.8 | 0.985 | 1.970; 1.975; 1.975 | 105.9; 96.5; 93.2 | 0.986 |
| 3.3 | 1.967 | 96.9 | 0.989 | 0.987 | |||
| 6.4 | 1.967 | 95.9 | 0.990 | 0.991 | |||
| 7.4 | 1.967 | 94.9 | 0.970 | 0.986 | |||
| 8.0 | 1.967 | 94.0 | 0.981 | 0.988 | |||
| S4 | 2.5 | 1.964; 1.968 | 104.4; 96.7 | 0.990 | 1.970; 1.974; 1.976 | 105.9; 96.1; 92.9 | 0.988 |
| 3.3 | 1.967 | 96.8 | 0.992 | 0.986 | |||
| 6.6 | 1.967 | 95.8 | 0.989 | 0.989 | |||
| 7.4 | 1.967 | 94.9 | 0.983 | 0.986 | |||
| 8.1 | 1.967 | 93.8 | 0.985 | 0.984 | |||
| SC | VO(H2O)5; VO(SC)H5 and 4 | VO(H2O)5; VO(SC)H5; VO(SC)H4 or 3 | |||||
| 2.8 | 1.967; 1.966 | 104.2; 97.6 | 0.990 | 1.970; 1.974; 1.976 | 105.9; 96.7; 92.7 | 0.985 | |
| 3.1 | 1.967 | 96.9 | 0.994 | 0.991 | |||
| 6.5 | 1.967 | 95.6 | 0.989 | 0.992 | |||
| 7.5 | 1.967 | 94.3 | 0.980 | 0.987 | |||
| 8.0 | 1.967 | 93.4 | 0.980 | 0.983 | |||
| Ligand | pH | g‖ | A‖ (cm−1 ∙ 10−4) | g⊥ | A⊥ (cm−1 ∙ 10−4) |
|---|---|---|---|---|---|
| H2O | 3.0 | 1.933 | 180.8 | 1.978 | 70.2 |
| S2 | 6.2 | 1.939 | 168.9 | 1.975 | 59.8 |
| 8.2 | 1.941 | 167.2 | 1.974 | 57.1 | |
| S3 | 6.4 | 1.936 | 169.9 | 1.975 | 59.9 |
| 8.2 | 1.939 | 167.1 | 1.972 | 56.3 | |
| S4 | 6.4 | 1.936 | 170.8 | 1.975 | 60.8 |
| 8.0 | 1.940 | 166.9 | 1.973 | 56.9 | |
| SC | 5.6 | 1.938 | 170.2 | 1.974 | 59.2 |
| 8.0 | 1.938 | 167.0 | 1.971 | 56.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berto, S.; Alladio, E.; Daniele, P.G.; Laurenti, E.; Bono, A.; Sgarlata, C.; Valora, G.; Cappai, R.; Lachowicz, J.I.; Nurchi, V.M. Oxovanadium(IV) Coordination Compounds with Kojic Acid Derivatives in Aqueous Solution. Molecules 2019, 24, 3768. https://doi.org/10.3390/molecules24203768
Berto S, Alladio E, Daniele PG, Laurenti E, Bono A, Sgarlata C, Valora G, Cappai R, Lachowicz JI, Nurchi VM. Oxovanadium(IV) Coordination Compounds with Kojic Acid Derivatives in Aqueous Solution. Molecules. 2019; 24(20):3768. https://doi.org/10.3390/molecules24203768
Chicago/Turabian StyleBerto, Silvia, Eugenio Alladio, Pier Giuseppe Daniele, Enzo Laurenti, Andrea Bono, Carmelo Sgarlata, Gabriele Valora, Rosita Cappai, Joanna Izabela Lachowicz, and Valeria Marina Nurchi. 2019. "Oxovanadium(IV) Coordination Compounds with Kojic Acid Derivatives in Aqueous Solution" Molecules 24, no. 20: 3768. https://doi.org/10.3390/molecules24203768