C–H-Bond Activation and Isoprene Polymerization Studies Applying Pentamethylcyclopentadienyl-Supported Rare-Earth-Metal Bis(Tetramethylaluminate) and Dimethyl Complexes

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Pentamethylcyclopentadienyl Lanthanide Bis(tetramethylaluminate) Compelxes
2.2. Ether-Promoted Methylaluminato Cleavage Reactions of [Cp*Ln(AlMe4)2] (1Ln)
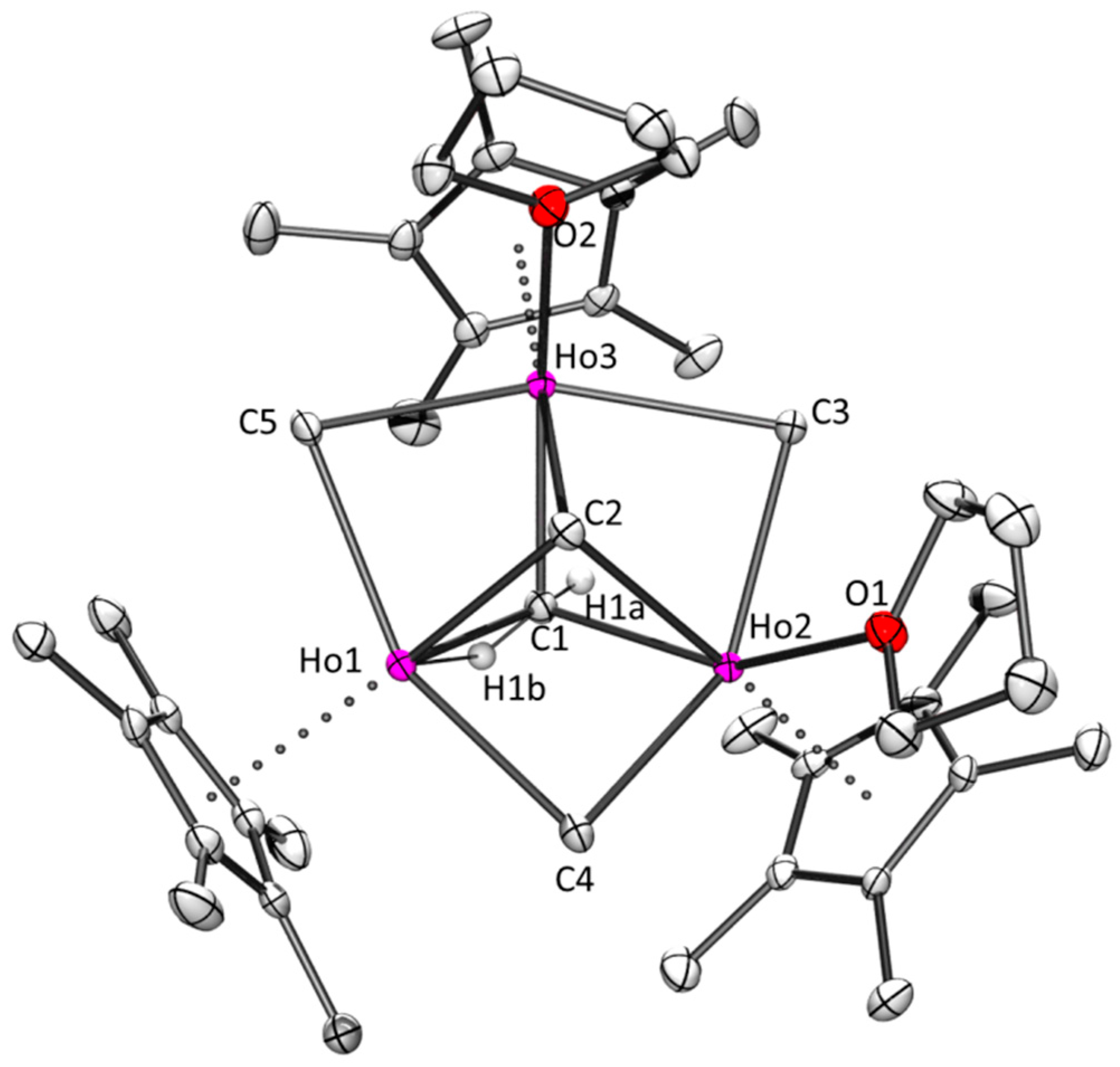
2.2.1. Holmium and Dysprosium
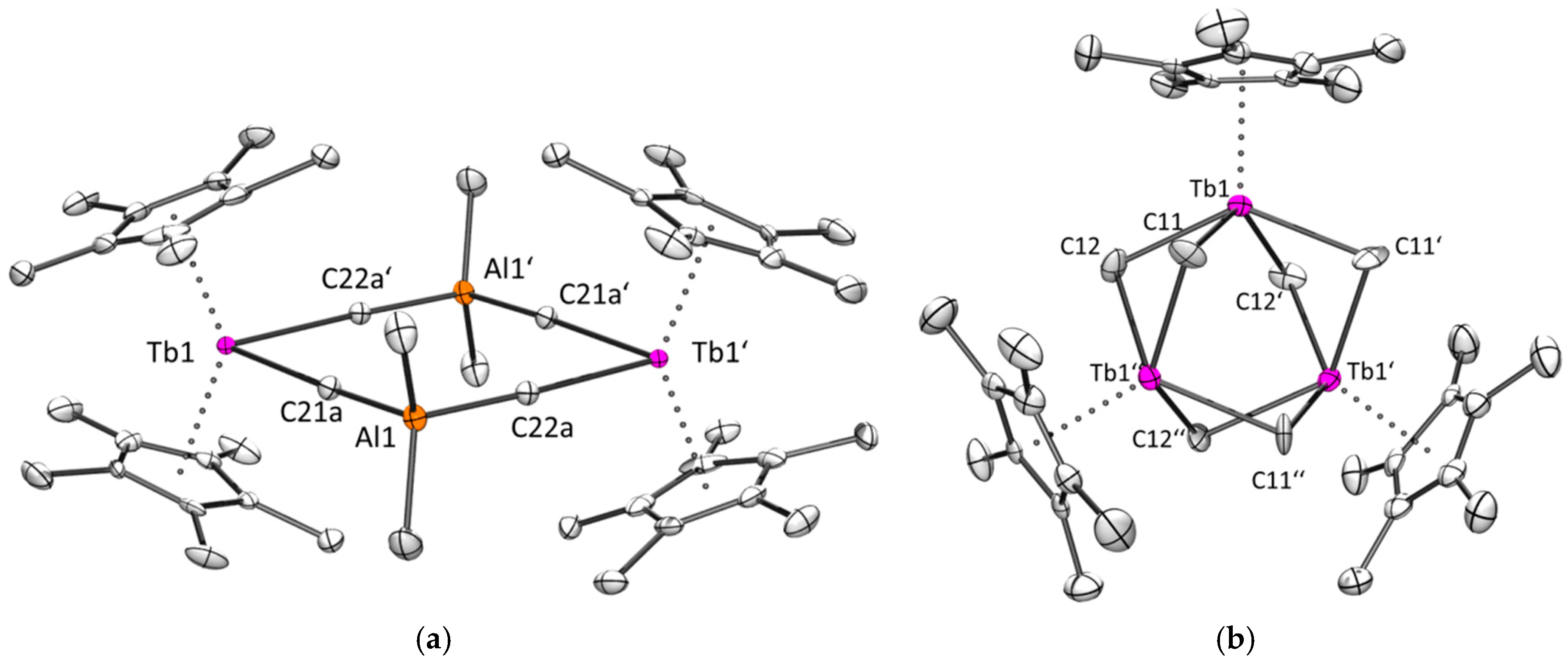
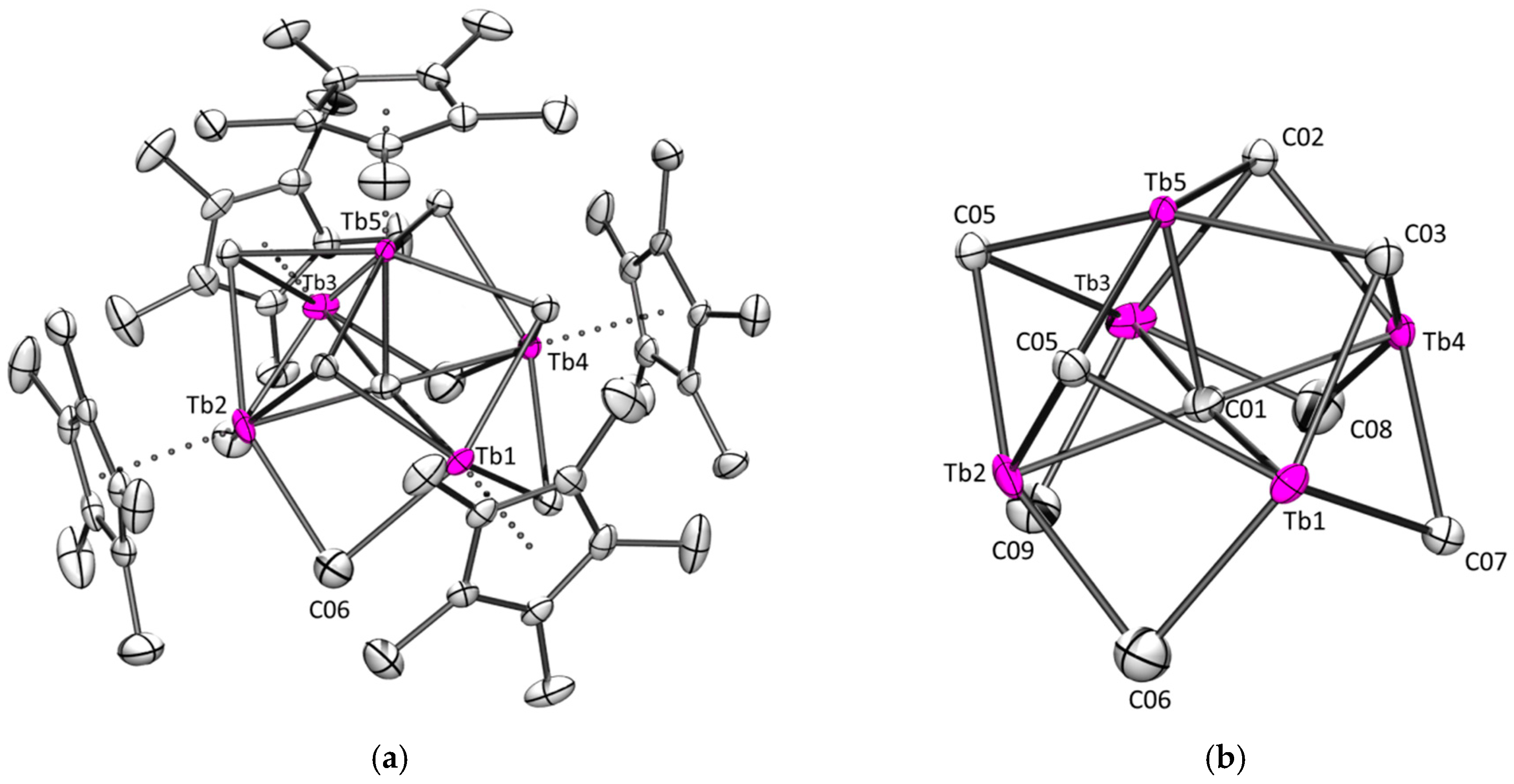
2.2.2. Terbium
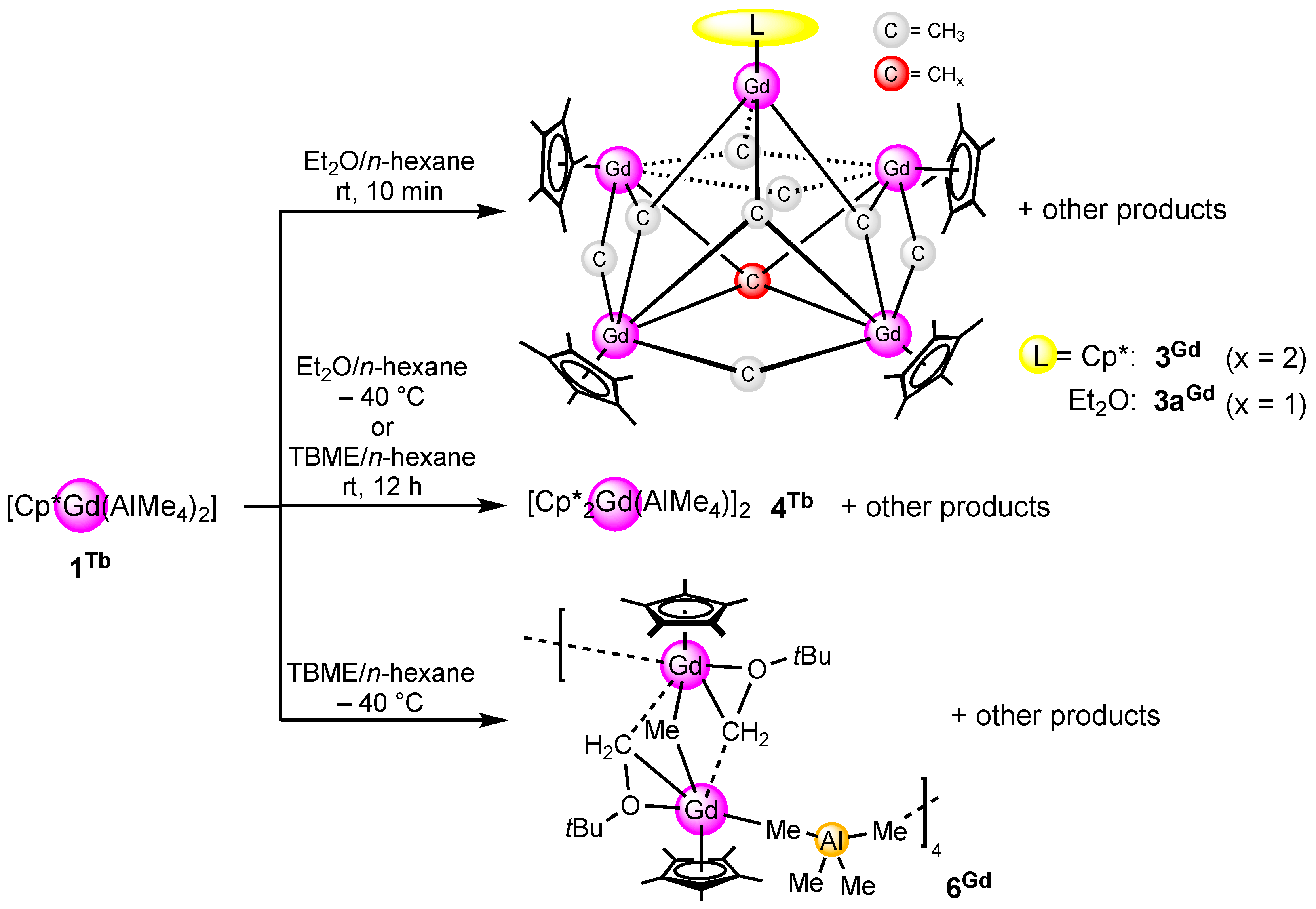
2.2.3. Gadolinium
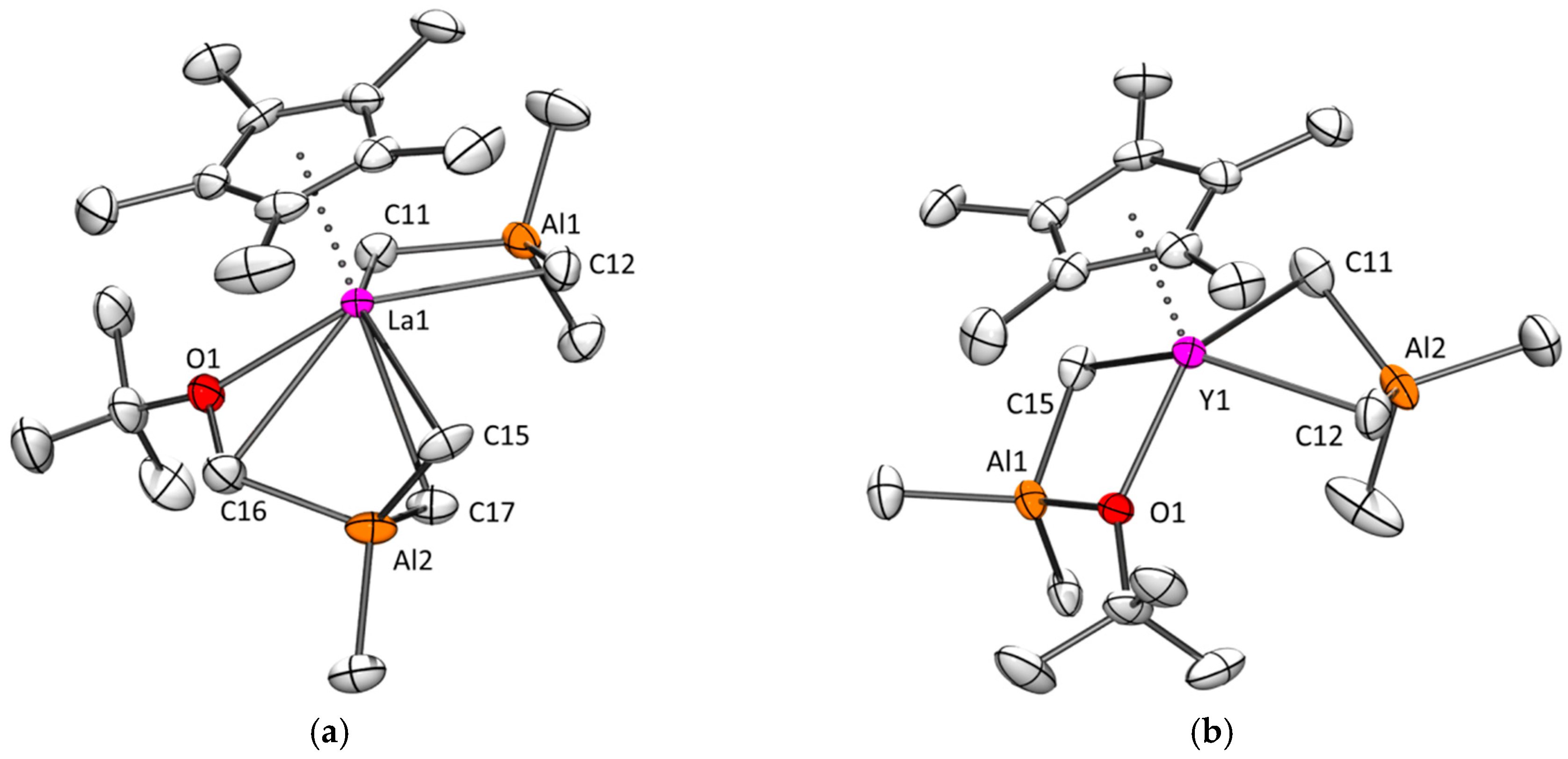
2.2.4. More about Yttrium and Lanthanum: Structural Snapshots
2.3. C–H-Bond Activation of [Cp*HoMe2]3 upon Reaction with THF
3. Materials and Methods
3.1. General Remarks
3.2. Synthesis of [Cp*Ho(AlMe4)2] (1Ho)
3.3. Synthesis of [Cp*Dy(AlMe4)2] (1Dy)
3.4. Synthesis of [Cp*Tb(AlMe4)2] (1Tb)
3.5. Synthesis of [Cp*Gd(AlMe4)2] (1Gd)
3.6. Synthesis of [Cp*HoMe2]3 (2Ho)
3.7. Synthesis of [Cp*DyMe2]3 (2Dy)
3.8. Synthesis of [Cp*TbMe2]3 (2Tb) and [(Cp*Tb)5(CH2)(Me)8] (3Tb)
3.9. Synthesis of [(Cp*Gd)5(CH2)(Me)8] (3Gd) and [(Cp*Gd)4((Et2O)Gd)(CH)(Me)8] (3aGd)
3.10. Synthesis of [Cp*2Tb(AlMe4)]2 (4Tb)
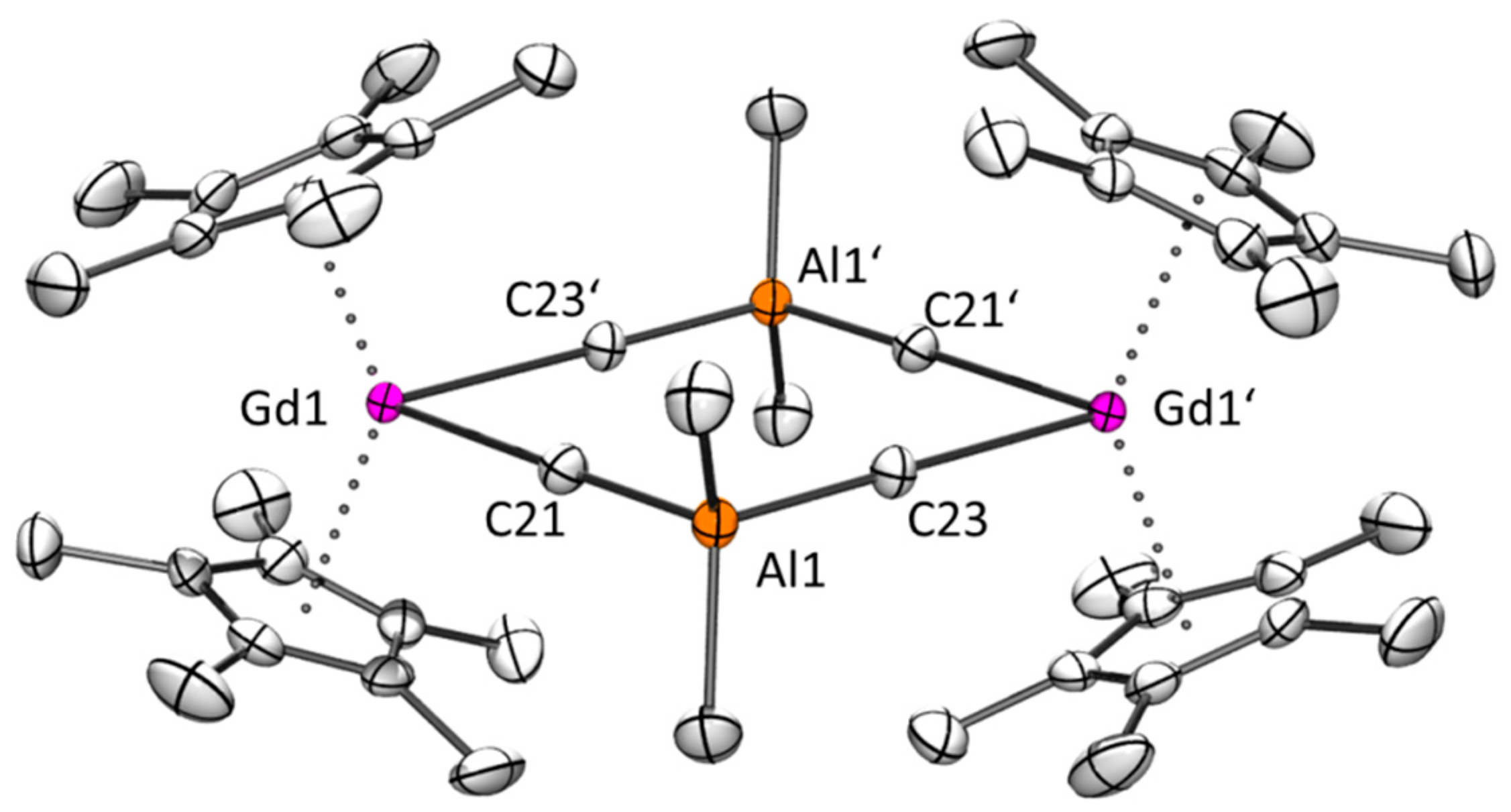
3.11. Synthesis of [Cp*2Gd(AlMe4)]2 (4Gd) According to Two Different Routes
3.12. Synthesis of [(Cp*Y)5(Me)6(CH2)2] (5Y)
3.13. Synthesis of [{(Cp*Gd)2(Me)(CH2OtBu)2(AlMe4)}2]2 (6Gd)
3.14. Synthesis of [Cp*La(AlMe4){Me3Al(CH2)OtBu}] (7La)
3.15. Synthesis of [Cp*Y(AlMe4)(Me3AlOtBu)] (8Y)
3.16. Synthesis of [(Cp*Ho)3Me4(CH2)(thf)2] (9Ho)
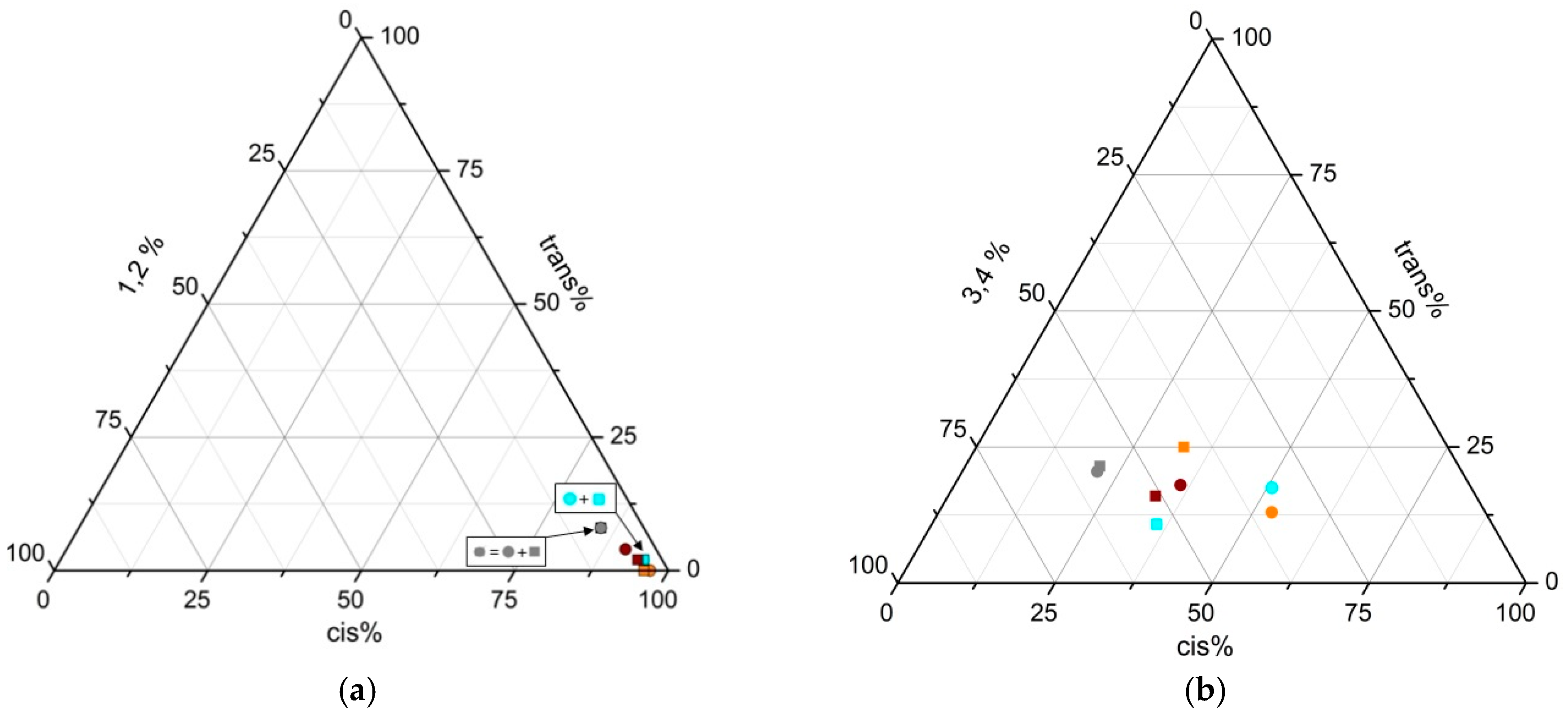
3.17. Polymerization of Isoprene
3.18. Polymerization of 1,3-Butadiene
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van der Heijden, H.; Schaverien, C.J.; Orper, A.G. The first salt- and solvent-free monocyclopentadienyl lanthanide dialkyl complex. X-ray structure determinations of La(η5-C5Me5)[CH(SiMe3)2]2 and of its tetrahydrofuran adduct: compounds containing agostic silicon-carbon bonds. Organometallics 1989, 8, 255–258. [Google Scholar] [CrossRef]
- Luo, Y.; Baldamus, J.; Hou, Z. Scandium Half-Metallocene-Catalyzed Syndiospecific Styrene Polymerization and Styrene−Ethylene Copolymerization: Unprecedented Incorporation of Syndiotactic Styrene−Styrene Sequences in Styrene−Ethylene Copolymers. J. Am. Chem. Soc. 2004, 126, 13910–13911. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Baldamus, J.; Hou, Z. Alternating Ethylene–Norbornene Copolymerization Catalyzed by Cationic Half-Sandwich Scandium Complexes. Angew. Chem. Int. Ed. 2005, 44, 962–965. [Google Scholar] [CrossRef] [PubMed]
- Hultzsch, K.C.; Spaniol, T.P.; Okuda, J. Half-Sandwich Alkyl and Hydrido Complexes of Yttrium: Convenient Synthesis and Polymerization Catalysis of Polar Monomers. Angew. Chem. Int. Ed. 1999, 38, 227–230. [Google Scholar] [CrossRef]
- Bambirra, S.; van Leusen, D.; Meetsma, A.; Hessen, B.; Teuben, J.H. Yttrium alkyl complexes with a sterically demanding benzamidinate ligand: synthesis, structure and catalytic ethene polymerization. Chem. Commun. 2003, 522–523. [Google Scholar] [CrossRef] [PubMed]
- Bambirra, S.; Bouwkamp, M.W.; Meetsma, A.; Hessen, B. Cationic Mono(amidinate) Alkyl Catalysts over the Full Size Range of the Group 3 and Lanthanide Metals. J. Am. Chem. Soc. 2004, 126, 9182–9183. [Google Scholar] [CrossRef]
- Arndt, S.; Okuda, J. Mono(cyclopentadienyl) Complexes of the Rare-Earth Metals. Chem. Rev. 2002, 102, 1953–1976. [Google Scholar] [CrossRef]
- Hou, Z.; Luo, Y.; Li, X. Cationic rare earth metal alkyls as novel catalysts for olefin polymerization and copolymerization. J. Organomet. Chem. 2006, 691, 3114–3121. [Google Scholar] [CrossRef]
- Rodrigues, A.-S.; Carpentier, J.-F. Groups 3 and 4 single site catalysts for styrene-ethylene and styrene-α-olefin copolymerization. Coord. Chem. Rev. 2008, 2137–2154. [Google Scholar] [CrossRef]
- Nishiura, M.; Guo, F.; Hou, Z. Half-Sandwich Rare-Earth-Catalyzed Olefin Polymerization, Carbometalation, and Hydroarylation. Acc. Chem. Res. 2015, 48, 2209–2220. [Google Scholar] [CrossRef]
- Anwander, R.; Klimpel, M.G.; Martin Dietrich, H.; Shorokhov, D.J.; Scherer, W. High tetraalkylaluminate fluxionality in half-sandwich complexes of the trivalent rare-earth metals. Chem. Commun. 2003, 1008–1009. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Törnroos, K.W.; Anwander, R. Cationic Rare-Earth-Metal Half-Sandwich Complexes for the Living trans-1,4-Isoprene Polymerization. Angew. Chem. Int. Ed. 2008, 47, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Törnroos, K.W.; Sitzmann, H.; Anwander, R. Half-Sandwich Bis(tetramethylaluminate) Complexes of the Rare-Earth Metals: Synthesis, Structural Chemistry, and Performance in Isoprene Polymerization. Chem. Eur. J. 2008, 14, 7266–7277. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Volbeda, J.; Törnroos, K.W.; Anwander, R. Tetramethylcyclopentadienyl-supported rare-earth metal bis(tetramethyl)aluminate complexes: Synthesis, structural chemistry, cation formation, and isoprene polymerization. C. Rend. Chim. 2010, 13, 651–660. [Google Scholar] [CrossRef]
- Thiele, S.; Wilson, D.R. Alternate Transition Metal Complex Based Diene Polymerization. J. Macromol. Sci. Part C 2004, 43, 581–628. [Google Scholar] [CrossRef]
- Friebe, L.; Nuyken, O.; Obrecht, W. Neodymium-Based Ziegler/Natta Catalysts and their Application in Diene Polymerization. Adv. Polym. Sci. 2006, 204, 1–154. [Google Scholar]
- Fischbach, A.; Anwander, R. Rare-Earth Metals and Aluminum getting Close in Ziegler Type Organometallics. Adv. Polym. Sci. 2006, 204, 155–281. [Google Scholar]
- Zhang, Z.; Cui, D.; Wang, B.; Liu, B.; Yang, Y. Molecular Catalysis of Rare-Earth Elements; Roesky, P.W., Ed.; Springer: Berlin, Germany, 2010; pp. 49–108. [Google Scholar]
- Kelly, R.P.; Roesky, P.W. Catalytic Sigma-Bond Metathesis and the Polymerization of 1,3-Dienes by Rare-Earth Metal Complexes. Struct Bond 2016, 172, 85–118. [Google Scholar]
- Jothieswaran, J.; Fadlallah, S.; Bonnet, F.; Visseaux, M. Recent Advances in Rare Earth Complexes Bearing Allyl Ligands and Their Reactivity towards Conjugated Dienes and Styrene Polymerization. Catalysts 2017, 7, 378. [Google Scholar] [CrossRef]
- Trifonov, A.A.; Lyubov, D.M. A quarter-century long story of bis(alkyl) rare-earth (III) complexes. Coord. Chem. Rev. 2017, 340, 10–61. [Google Scholar] [CrossRef]
- Huang, J.; Liu, Z.; Cui, D.; Liu, X. Precisely Controlled Polymerization of Styrene and Conjugated Dienes by Group 3 Single-Site Catalysts. ChemCatChem 2018, 10, 42–61. [Google Scholar] [CrossRef]
- Dietrich, H.M.; Grove, H.; Törnroos, K.W.; Anwander, R. Multiple C−H Bond Activation in Group 3 Chemistry: Synthesis and Structural Characterization of an Yttrium−Aluminum−Methine Cluster. J. Am. Chem. Soc. 2006, 128, 1458–1459. [Google Scholar] [CrossRef] [PubMed]
- Hollfelder, C.O.; Jende, L.N.; Dietrich, H.-M.; Eichele, K.; Maichle-Mössmer, C.; Anwander, R. 1,3-Diene Polymerization Promoted by Half-Sandwich Rare-Earth-Metal Dimethyl Complexes: Active Species Clustering and Cationization/Deactivation Processes. Chem. Eur. J. 2019, 25, 7298–7302. [Google Scholar] [CrossRef] [PubMed]
- Kaita, S.; Hou, Z.; Nishiura, M.; Doi, Y.; Kurazumi, J.; Horiuchi, A.C.; Wakatsuki, Y. Ultimately Specific 1,4-cis Polymerization of 1,3-Butadiene with a Novel Gadolinium Catalyst. Macromol. Rapid Commun. 2003, 24, 179–184. [Google Scholar] [CrossRef]
- Kaita, S.; Yamanaka, M.; Horiuchi, A.C.; Wakatsuki, Y. Butadiene Polymerization Catalyzed by Lanthanide Metallocene−Alkylaluminum Complexes with Cocatalysts: Metal-Dependent Control of 1,4-Cis/Trans Stereoselectivity and Molecular Weight. Macromolecules 2006, 39, 1359–1363. [Google Scholar] [CrossRef]
- Fischbach, A.; Klimpel, M.G.; Widenmeyer, M.; Herdtweck, E.; Scherer, W.; Anwander, R. Stereospecific Polymerization of Isoprene with Molecular and MCM-48-Grafted Lanthanide(III) Tetraalkylaluminates. Angew. Chem. Int. Ed. 2004, 43, 2234–2239. [Google Scholar] [CrossRef] [PubMed]
- Meermann, C.; Törnroos, K.W.; Nerdal, W.; Anwander, R. Rare-Earth Metal Mixed Chloro/Methyl Compounds: Heterogeneous–Homogeneous Borderline Catalysts in 1,3-Diene Polymerization. Angew. Chem. Int. Ed. 2007, 46, 6508–6513. [Google Scholar] [CrossRef]
- Shannon, R. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Shannon, R.D.; Prewitt, C.T. Effective ionic radii in oxides and fluorides. Acta Cryst. B 1969, 25, 925–946. [Google Scholar] [CrossRef]
- Evans, W.J.; Anwander, R.; Ziller, J.W. Inclusion of Al2Me6 in the Crystalline Lattice of the Organometallic Complexes LnAl3Me12. Organometallics 1995, 14, 1107–1109. [Google Scholar] [CrossRef]
- Klooster, W.T.; Lu, R.S.; Anwander, R.; Evans, W.J.; Koetzle, T.F.; Bau, R. Neutron Diffraction Study of [Nd(AlMe4)3] 0.5 Al2Me6 at 100 K: The First Detailed Look at a Bridging Methyl Group with a Trigonal-Bipyramidal Carbon Atom. Angew. Chem. Int. Ed. 1998, 37, 1268–1270. [Google Scholar] [CrossRef]
- Zimmermann, M.; Frøystein, N.Å.; Fischbach, A.; Sirsch, P.; Dietrich, H.M.; Törnroos, K.W.; Herdtweck, E.; Anwander, R. Homoleptic Rare-Earth Metal(III) Tetramethylaluminates: Structural Chemistry, Reactivity, and Performance in Isoprene Polymerization. Chem. Eur. J. 2007, 13, 8784–8800. [Google Scholar] [CrossRef] [PubMed]
- Occhipinti, G.; Meermann, C.; Dietrich, H.M.; Litlabø, R.; Auras, F.; Törnroos, K.W.; Maichle-Mössmer, C.; Jensen, V.R.; Anwander, R. Synthesis and Stability of Homoleptic Metal(III) Tetramethylaluminates. J. Am. Chem. Soc. 2011, 133, 6323–6337. [Google Scholar] [CrossRef] [PubMed]
- Nieland, A.; Mix, A.; Neumann, B.; Stammler, H.G.; Mitzel, N.W. Lanthanoid Tetramethylaluminates and Their Paramagnetic NMR Parameters. Eur. J. Inorg. Chem. 2014, 2014, 51–57. [Google Scholar] [CrossRef]
- König, S.N.; Chilton, N.F.; Maichle-Mössmer, C.; Pineda, E.M.; Pugh, T.; Anwander, R.; Layfield, R.A. Fast magnetic relaxation in an octahedral dysprosium tetramethyl-aluminate complex. Dalton Trans. 2014, 43, 3035–3038. [Google Scholar] [CrossRef] [PubMed]
- Hollfelder, C.O.; Jende, L.N.; Diether, D.; Zelger, T.; Stauder, R.; Maichle-Mössmer, C.; Anwander, R. 1,3-Diene Polymerization Mediated by Homoleptic Tetramethylaluminates of the Rare-Earth Metals. Catalysts 2018, 8, 61. [Google Scholar] [CrossRef]
- Zimmermann, M.; Anwander, R. Homoleptic Rare-Earth Metal Complexes Containing Ln−C σ-Bonds. Chem. Rev. 2010, 110, 6194–6259. [Google Scholar] [CrossRef]
- Dietrich, H.M.; Zapilko, C.; Herdtweck, E.; Anwander, R. Ln(AlMe4)3 as New Synthetic Precursors in Organolanthanide Chemistry: Efficient Access to Half-Sandwich Hydrocarbyl Complexes. Organometallics 2005, 24, 5767–5771. [Google Scholar] [CrossRef]
- Dietrich, H.M.; Törnroos, K.W.; Herdtweck, E.; Anwander, R. Tetramethylaluminate and Tetramethylgallate Coordination in Rare-Earth Metal Half-Sandwich and Metallocene Complexes. Organometallics 2009, 28, 6739–6749. [Google Scholar] [CrossRef]
- Litlabø, R.; Enders, M.; Törnroos, K.W.; Anwander, R. Bis(tetramethylaluminate) Complexes of Yttrium and Lanthanum Supported by a Quinolyl-Substituted Cyclopentadienyl Ligand: Synthesis and Performance in Isoprene Polymerization. Organometallics 2010, 29, 2588–2595. [Google Scholar] [CrossRef]
- Dettenrieder, N.; Hollfelder, C.O.; Jende, L.N.; Maichle-Mössmer, C.; Anwander, R. Half-Sandwich Rare-Earth-Metal Alkylaluminate Complexes Bearing Peripheral Boryl Ligands. Organometallics 2014, 33, 1528–1531. [Google Scholar] [CrossRef]
- Bienfait, A.M.; Wolf, B.M.; Törnroos, K.W.; Anwander, R. Ln(II)/Pb(II)–Ln(III)/Pb(0) Redox Approach toward Rare-Earth-Metal Half-Sandwich Complexes. Organometallics 2015, 34, 5734–5744. [Google Scholar] [CrossRef]
- Holton, J.; Lappert, M.F.; Ballard, D.G.H.; Pearce, R.; Atwood, J.L.; Hunter, W.E. Alkyl-bridged complexes of the d- and f-block elements. Part 2. Bis[bis(η-cyclopentadienyl)methylmetal(III)] complexes, and the crystal and molecular structure of the yttrium and ytterbium species. J. Chem. Soc. Dalton Trans. 1979, 54–61. [Google Scholar] [CrossRef]
- Zhang, W.X.; Wang, Z.; Nishiura, M.; Zhenfeng, X.; Hou, Z. Ln4(CH2)4 Cubane-Type Rare-Earth Methylidene Complexes Consisting of “(C5Me4SiMe3)LnCH2” Units (Ln = Tm, Lu). J. Am. Chem. Soc. 2011, 133, 5712–5715. [Google Scholar] [CrossRef]
- Jian, Z.; Cui, D.; Hou, Z.; Li, X. Living catalyzed-chain-growth polymerization and block copolymerization of isoprene by rare-earth metal allyl precursors bearing a constrained-geometry-conformation ligand. Chem. Commun. 2010, 46, 3022–3024. [Google Scholar] [CrossRef]
- Ajellal, N.; Furlan, L.; Thomas, C.M.; Casagrande, O.L.; Carpentier, J.-F. Mixed Aluminum-Magnesium-Rare Earth Allyl Catalysts for Controlled Isoprene Polymerization: Modulation of Stereocontrol. Macromol. Rapid Commun. 2006, 27, 338–343. [Google Scholar] [CrossRef]
- Valente, A.; Mortreux, A.; Visseaux, M.; Zinck, P. Coordinative Chain Transfer Polymerization. Chem. Rev. 2013, 113, 3836–3857. [Google Scholar] [CrossRef]
- Zhang, L.; Nishiura, M.; Yuki, M.; Luo, Y.; Hou, Z. Isoprene Polymerization with Yttrium Amidinate Catalysts: Switching the Regio- and Stereoselectivity by Addition of AlMe3. Angew. Chem. Int. Ed. 2008, 47, 2642–2645. [Google Scholar] [CrossRef]
- Jende, L.N.; Hollfelder, C.O.; Maichle-Mössmer, C.; Anwander, R. Rare-Earth-Metal Allyl Complexes Supported by the [2-(N,N-Dimethylamino)ethyl]tetramethylcyclopentadienyl Ligand: Structural Characterization, Reactivity, and Isoprene Polymerization. Organometallics 2015, 34, 32–41. [Google Scholar] [CrossRef]
- Busch, M.A.; Harlow, R.; Watson, P.L. Unusual eight-membered rings with linear symmetric methyl groups from yttrium and lutetium methyl complexes. Inorg. Chim. Acta 1987, 140, 15–20. [Google Scholar] [CrossRef]
- Evans, W.J.; Chamberlain, L.R.; Ulibarri, T.A.; Ziller, J.W. Reactivity of trimethylaluminum with (C5Me5)2Sm(THF)2: synthesis, structure, and reactivity of the samarium methyl complexes (C5Me5)2Sm[(µ-Me)AlMe2(µ-Me)]2Sm(C5Me5)2 and (C5Me5)2SmMe(THF). J. Am. Chem. Soc. 1988, 110, 6423–6432. [Google Scholar] [CrossRef]
- Takenaka, Y.; Shima, Y.; Baldemus, J.; Hou, Z. Reduction of Transition-Metal-Coordinated Carbon Monoxide by a Rare-Earth Hydride Cluster: Isolation of Well-Defined Heteromultimetallic Oxycarbene, Oxymethyl, Carbene, and Methyl Complexes. Angew. Chem. Int. Ed. 2009, 48, 7888–7891. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Nishiura, M.; Cheng, J.; Zhang, W.; Li, Y.; Hou, Z. Hydrogenolysis and Protonation of Polymetallic Lutetium Methylidene and Methyl Complexes. Organometallics 2013, 32, 4142–4148. [Google Scholar] [CrossRef]
- Dietrich, H.M.; Törnroos, K.W.; Anwander, R. “Ionic Carbenes”: Synthesis, Structural Characterization, and Reactivity of Rare-Earth Metal Methylidene Complexes. J. Am. Chem. Soc. 2006, 128, 9298–9299. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Boyle, T.J.; Ziller, J.W. New coordination environments for yttrium formed in situ by heterometallic bridging: Crystal structures of (C5H4SiMe3)Y[(μ-OCMe3)(μ-Me)AlMe2]2 and (Me3SiCH2)Y[(μ-CH2)2 SiMe2][(μ-OR)Li(THF)2]2. J. Organomet. Chem. 1993, 462, 141–148. [Google Scholar] [CrossRef]
- Fischbach, A.; Herdtweck, E.; Anwander, R. Synthesis and Derivatization of Halflanthanidocene Aryl(Alk)oxide Complexes. Inorg. Chim. Acta 2006, 359, 4855–4864. [Google Scholar] [CrossRef]
- COSMO v. 1.61; Bruker AXS Inc.: Madison, WI, USA, 2012.
- APEX 3 V. 2017.3-0; Bruker AXS Inc.: Madison, WI, USA, 2016.
- SAINT v. 8.38A; Bruker AXS Inc.: Madison, WI, USA, 2015.
- Sheldrick, G. SHELXT–Integrated space-group and crystal-structure determination. Acta Cryst. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl.Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Cryst. 2011, 44, 1281–1284. [Google Scholar] [CrossRef]
- Kratzert, D.; Holstein, J.J.; Krossing, I. DSR: Enhanced modelling and refinement of disordered structures with SHELXL. J. Appl. Cryst. 2015, 48, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. ORTEP-3 for Windows—a version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- POV-Ray, Version 3.6; Vision Pty. Ltd.: Williamstown, Australia, 2004.
Sample Availability: Samples of the compounds are not available from the authors. |
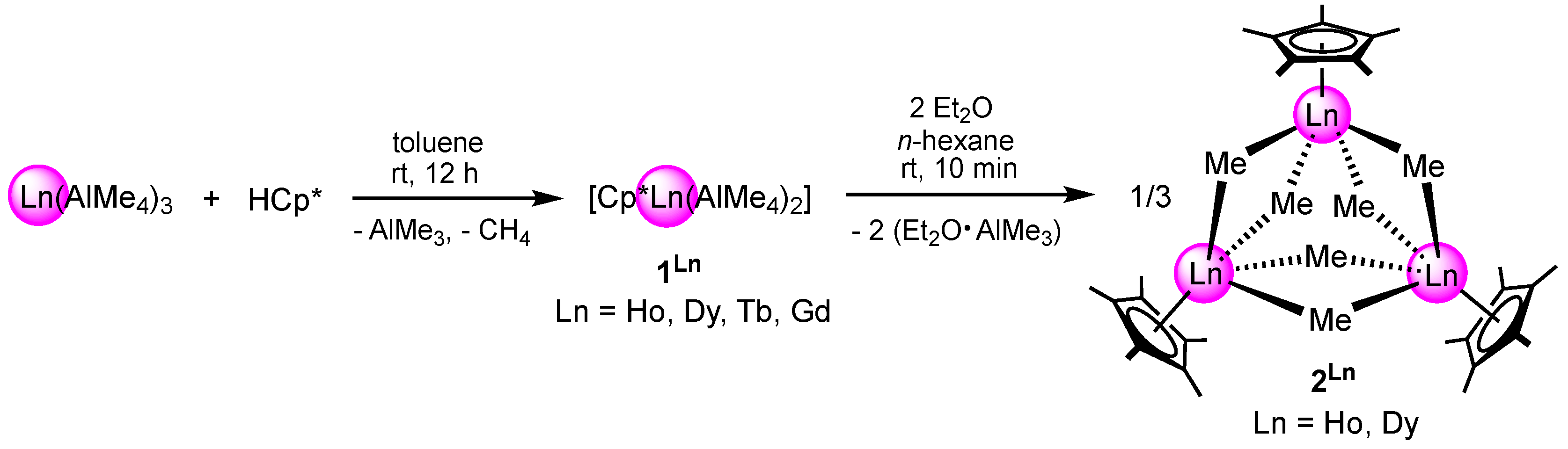










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ln | ø(Ln–CAl)a | Ln–Ct | ø(∠ Ct–Ln–CAl)a | Ln---C14 a | ø(∠ CAl1–Ln–CAl1)a | ø(∠ CAl1–Ln–CAl2)a | T1a | T2a |
|---|---|---|---|---|---|---|---|---|
| (Å) | (Å) | (°) | (Å) | (°) | (°) | (°) | (°) | |
| Lab | 2.749(4) | 2.503 | 106.9 | 3.140 | 75.7(1) | 94.4(1) | 2.2 | 32.5 |
| Smc | 2.668(2) | 2.404 | 106.9 | 3.113(2) | 78.19(6) | 91.41(6) | 3.5 | 31.3 |
| Gd | 2.637(2) | 2.376 | 107.4 | 3.163(2) | 79.59(7) | 90.05(7) | 4.1 | 30.7 |
| Tb | 2.625(2) | 2.362 | 107.4 | 3.169(2) | 80.15(7) | 89.27(7) | 4.4 | 30.6 |
| Dy | 2.608(2) | 2.344 | 107.7 | 3.186(2) | 80.46(5) | 88.78(6) | 4.7 | 30.2 |
| Ho | 2.602(4) | 2.334 | 108.1 | 3.248(4) | 80.8(1) | 88.0(1) | 5.1 | 29.9 |
| Yd | 2.601(3) | 2.340 | 108.6 | 3.304 | 80.5(1) | 87.7(1) | 5.4 | 29.4 |
| Ybc | 2.563(2) | 2.293 | 109.1 | 3.354(2) | 81.92(5) | 85.63(5) | 6.1 | 28.9 |
| Lue | 2.545(3) | 2.286 | 109.6 | 3.447 | 82.2(1) | 84.7(1) | 6.4 | 28.3 |
| Run a | Precata-lyst | Cocata-lyst b | Tc | Tc | Yield d | cis-1,4 e | trans-1,4 e | 3,4e | Mnf | PDIf | Tgg |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (°C) | (h) | (%) | (%) | (%) | (%) | (104 g·mol−1) | (°C) | ||||
| 1i | 1La | A | 40 | 24 | >99 | 4 | 87 | 9 | 7 | 1.28 | n.d.k |
| 2i | 1La | B | 40 | 24 | >99 | 3 | 80 | 17 | 6 | 1.22 | n.d.k |
| 3i | 1La | C | 40 | 24 | >99 | 0 | 99.5 | 0.5 | 24 | 1.18 | n.d.k |
| 4i | 1Nd | A | 40 | 24 | >99 | 14 | 70 | 16 | 3 | 2.87 | n.d.k |
| 5i | 1Nd | B | 40 | 24 | >99 | 7 | 80 | 13 | 4 | 1.16 | n.d.k |
| 6i | 1Nd | C | 40 | 24 | >99 | 4 | 92 | 4 | 13 | 1.35 | n.d.k |
| 7 | 1Gd | A | 25 | 1 | 93 | 16 | 68 | 16 | 10.6 | 1.33 | −57 |
| 8 | 1Gd | B | 25 | 1 | 87 | 22 | 45 | 33 | 4.3 | 1.19 | −43 |
| 9 | 1Gd | C | 25 | 1 | traces h | - h | - h | - h | - h | - h | - h |
| 10 | 1Gd | C | 25 | 24 | 94 | 6 | 87 | 7 | 19.4 | 1.30 | −64 |
| 11 | 1Tb | A | 25 | 1 | 89 | 15 | 66 | 19 | 9.9 | 1.53 | −49 |
| 12 | 1Tb | B | 25 | 1 | 82 | 25 | 34 | 41 | 3.5 | 1.58 | −53 |
| 13 | 1Tb | C | 25 | 1 | 33 | 32 | 62 | 6 | 7.8 | 1.87 | −62 |
| 14 | 1Dy | A | 25 | 1 | 94 | 20 | 43 | 37 | 12.7 | 1.31 | −52 |
| 15 | 1Dy | B | 25 | 1 | 83 | 14 | 63 | 23 | 5.0 | 1.28 | −43 |
| 16 | 1Dy | C | 25 | 1 | 11 | 39 | 52 | 9 | 3.7 | 1.54 | −65 |
| 17 | 1Ho | A | 25 | 1 | 54 | 23 | 50 | 27 | 11.0 | 1.50 | −38 |
| 18 | 1Ho | B | 25 | 1 | 81 | 29 | 24 | 47 | 4.1 | 1.48 | −23 |
| 19 | 1Ho | C | 25 | 1 | traces h | - h | - h | - h | - h | - h | - h |
| 20 | 1Ho | C | 25 | 24 | 85 | 9 | 79 | 12 | 6.6 | 1.62 | −62 |
| 21i | 1Y | A | 40 | 24 | >99 | 61 | 21 | 19 | 2 | 8.95 | n.d.k |
| 22i | 1Y | B | 40 | 24 | >99 | 44 | 29 | 28 | 6 | 1.59 | n.d.k |
| 23i | 1Y | C | 40 | 24 | >99 | 2 | 94 | 4 | 9 | 1.59 | n.d.k |
| 24j | 1Lu | A | 40 | 0.25 | >99 | 74 | 20 | 6 | 10 | 1.49 | n.d.k |
| 25j | 1Lu | B | 40 | 0.25 | >99 | 70 | 20 | 10 | 9.5 | 1.48 | n.d.k |
| 26j | 1Lu | C | 40 | 0.25 | >99 | 74 | 21 | 5 | 11.0 | 1.39 | n.d.k |
| Ln | Ln–C11 | Ln–C12 | Ln–Ct | ø (∠ Ct–Ln–C11/C12) | ∠ C11–Ln–C12 | ∠ C11–Ln–C11’’/∠ C12–Ln–C12’’ |
|---|---|---|---|---|---|---|
| (Å) | (Å) | (Å) | (°) | (°) | (°) | |
| Tb | 2.54(2) | 2.57(2) | 2.389 | 114.0 | 79.8(3) | 83(1)/83.9(7) |
| Dy | 2.63(3) | 2.49(3) | 2.360 | 111.6 | 81(1) | 85.1(9)/79.9(3) |
| Ho | 2.58(2) | 2.58(2) | 2.399 | 112.7 | 80.7(3) | 77.5(7)/87.2(8) |
| Y a | 2.539 | - a | 2.359 | 112.9 | 80.0 | 82.8 a |
| Lu b | 2.45(2) | 2.53(3) | 2.317 | 111.9 | 80.4(3) | 88(1)/79 (1) |
| Run a | Precata-lyst | Cocata-lyst b | Tc | tc | Yield d | cis-1,4 e | trans-1,4 e | 1,2 e | Mnf | PDI f | Tgg |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (°C) | (h) | (%) | (%) | (%) | (%) | (104 g·mol−1) | (°C) | ||||
| 27 | 2Dy | A | 25 | 60 | 99 | 97 | 0 | 3 | 16.2 | 1.63 | −102 |
| 28 | 2Dy | B | 25 | 60 | 92 | 96 | 0 | 4 | 20.4 | 1.64 | −102 |
| 29 | 2Ho | A | 25 | 60 | 90 | 91 | 4 | 5 | 5.5 | 1.52 | −102 |
| 30 | 2Ho | B | 25 | 60 | 59 | 94 | 2 | 4 | 32.8 | 1.35 | −101 |
| 31j | 2Y | A | 25 | 60 | 21 | 91 | 4 | 5 | 76 | 1.58 | −97 |
| 32j | 2Y | B | 25 | 60 | 48 | 95 | 2 | 3 | 50 | 1.20 | −94 |
| 33j | 2Lu | A | 25 | 60 | >99 | 85 | 8 | 7 | 40 | 1.84 | −94 |
| 34j | 2Lu | B | 25 | 60 | 95 | 85 | 8 | 7 | 33 | 1.77 | −93 |
| Runa | Precata-lyst | Cocata-lyst b | Tc | tc | Yield d | cis-1,4 e | trans-1,4 e | 3,4 e | Mnf | PDI f | Tgg |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (°C) | (h) | (%) | (%) | (%) | (%) | (104 g·mol−1) | (°C) | ||||
| 35 | 2Dy | A | 25 | 60 | 90 | 53 | 13 | 34 | 6.3 | 3.15 | −36 |
| 36 | 2Dy | B | 25 | 60 | 89 | 33 | 25 | 43 | 14.6 | 1.38 | −24 |
| 37 | 2Ho | A | 25 | 60 | 96 | 36 | 18 | 46 | 15.5 | 1.27 | −20 |
| 38 | 2Ho | B | 25 | 60 | 87 | 33 | 16 | 51 | 12.0 | 1.13 | −20 |
| 39j | 2Y | A | 25 | 60 | >99 | 51 | 17 | 32 | 8.4 | 2.33 | −47 |
| 40j | 2Y | B | 25 | 60 | 90 | 36 | 11 | 53 | 14.1 | 1.16 | −27 |
| 41 j | 2Lu | A | 25 | 60 | >99 | 21 | 21 | 58 | 26.8 | 2.00 | −16 |
| 42 j | 2Lu | B | 25 | 60 | >99 | 21 | 22 | 57 | 29.2 | 1.38 | −17 |
| Ln | ø(Ln–CAl) a | Ln–Ct1 | Ln–Ct2 | ø(∠ Ct–Ln–CAl) a | ∠ Ct1–Ln–Ct2 | ∠ CAl1–Ln–CAl1’ |
|---|---|---|---|---|---|---|
| (Å) | (Å) | (Å) | (°) | (°) | (°) | |
| La b | 2.848(2) | 2.522 | 2.516 | 104.5 | 139.62 | 87.36(4) |
| La(Tol) b,c | 2.867(4) | 2.527 | 2.513 | 104.3 | 140.04 | 87.7(1) |
| Smd | 2.75(2) | 2.414 | 2.419 | 105.1 | 138.6 | 85.0(5) |
| Sm(Tol) c,e | 2.749(4) | 2.421 | 2.424 | 105.8 | 135.7 | 87.7(1) |
| Gd | 2.712(2) | 2.399 | 2.400 | 104.9 | 138.5 | 86.95(6) |
| Tb | 2.690(2) | 2.385 | 2.380 | 104.4 | 139.7 | 86.9 |
| Y f | 2.66 | n.d. | n.d. | n.d. | n.d. | 84.9(7) |
| Yb(Tol) c,e | 2.634(2) | 2.314 | 2.326 | 105.0 | 138.1 | 87.12(6) |
| Ln–CCp* | Ln1-4–Ct1-4 | Ln5–Ct5/O | Ln1-4–µ2C | Ln5–µ3C | Ln1-4–µ3C | C01–Ln5 | C01–Ln1-4 | |||||
| (Å) | (Å) | (Å) | (Å) | (Å) | (Å) | (Å) | (Å) | |||||
| 3Tb | 2.701(4)–2.808(5) | 2.443–2.448. | 2.495 | 2.569(5)–2.658(6) | 2.610(5)–2.657(5) | 2.663(5)–2.774(5) | 2.493(4) | 2.462(5)–2.529(5) | ||||
| 3Gd | 2.69(1)–2.816(5) | 2.450–2.471 | 2.497 | 2.533(6)–2.571(6) | 2.593(7)–2.633(6) | 2.631(6)–2.677(6) | 2.373(7) | 2.494(7)–2.533(7) | ||||
| 3aGd | 2.686(8)–2.923 | 2.504–2.595 | 2.487 | 2.569(9)–2.629(8) | 2.571(8)–2.582(8) | 2.811(8)–2.898(8) | 2.222(7) | 2.506(7)–2.531(7) | ||||
| 5Y | 2.64(1)–2.685(8) | 2.372–2.391 | - | 2.494(9)–2.78(1) | 2.477(8)–2.711(8) | 2.399(9)–2.638(9) | - | - | ||||
| ∠(C01–Ln1-4–µ2C) | ∠(C01–Ln5–µ3C) | ∠(µ3Cx–Ln5µ3Cx+2) | ∠(Ln1-4–C01–Ln5) | ∠(Ln1/3–C01–Ln2/4) | ||||||||
| (°) | (°) | (°) | (°) | (°) | ||||||||
| 3Tb | 74.97(1)–78.0(2) | 77.6(2)–78.1(1) | 156.8(2)/155.6(2) | 87.4(1)–90.5(1) | 175.7(2)/176.4(2) | |||||||
| 3Gd | 76.0(2)–78.1(2) | 73.2(2)–74.2(2) | 147.3(2)–147.4(2) | 92.9(2)–96.2(2) | 170.9(3)/171.1(3) | |||||||
| 3aGd | 77.3(3)–78.9(2) | 85.4(3)–87.1(4) | 172.5(3)/173.2(3) | 89.9(3)–90.6(2) | 179.3(3)/178.8(4) | |||||||
| 5Y | - | - | 134.7(3)/135.2(4) | - | - | |||||||
| Run a | Precata-lyst | Cocata-lyst b | Tc | tc | Yield d | cis-1,4 e | trans-1,4 e | 3,4 e | Mnf | PDI f | Tgg |
|---|---|---|---|---|---|---|---|---|---|---|---|
| [°C] | [h] | [%] | [%] | [%] | [%] | [104 g·mol−1] | [°C] | ||||
| 43 | 9Ho | A | 25 | 1 | 71 h | 48 h | 16h | 36h | 13.2 h | 1.34h | −36h |
| 44 | 9Ho | B | 25 | 1 | 9 | 74 | 1 | 25 | 22.0 | 1.60 | −49 |
| 45i | 9Y | A | 25 | 1 | 48 | 44 | 14 | 41 | 13.9h | 1.42h | −31 |
| 46i | 9Y | B | 25 | 1 | 7 | 76 | 0 | 24 | 55.1 | 1.97 | −48 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hollfelder, C.O.; Meermann-Zimmermann, M.; Spiridopoulos, G.; Werner, D.; Törnroos, K.W.; Maichle-Mössmer, C.; Anwander, R. C–H-Bond Activation and Isoprene Polymerization Studies Applying Pentamethylcyclopentadienyl-Supported Rare-Earth-Metal Bis(Tetramethylaluminate) and Dimethyl Complexes. Molecules 2019, 24, 3703. https://doi.org/10.3390/molecules24203703
Hollfelder CO, Meermann-Zimmermann M, Spiridopoulos G, Werner D, Törnroos KW, Maichle-Mössmer C, Anwander R. C–H-Bond Activation and Isoprene Polymerization Studies Applying Pentamethylcyclopentadienyl-Supported Rare-Earth-Metal Bis(Tetramethylaluminate) and Dimethyl Complexes. Molecules. 2019; 24(20):3703. https://doi.org/10.3390/molecules24203703
Chicago/Turabian StyleHollfelder, Christoph O., Melanie Meermann-Zimmermann, Georgios Spiridopoulos, Daniel Werner, Karl W. Törnroos, Cäcilia Maichle-Mössmer, and Reiner Anwander. 2019. "C–H-Bond Activation and Isoprene Polymerization Studies Applying Pentamethylcyclopentadienyl-Supported Rare-Earth-Metal Bis(Tetramethylaluminate) and Dimethyl Complexes" Molecules 24, no. 20: 3703. https://doi.org/10.3390/molecules24203703