4. Materials and Methods
Infrared (IR) spectra were obtained using a Perkin Elmer 1720X FT-IR spectrometer (Perkin Elmer, Wattham, MA, USA). HRMS was performed using a JEOL JMS-700 (2) mass spectrometer (JEOL, Tokyo, Japan). NMR spectra were recorded at 27 °C using Agilent 300, 400-MR-DD2, and 600-DD2 spectrometers in CDCl3 using tetramethylsilane (TMS) as the internal standard. Liquid column chromatography was conducted using silica gel BW127ZH (Fuji Silysia Chemical Ltd., Tokyo, Japn). Analytical and preparative thin layer chromatography (TLC) analyses were performed using pre-coated Merck glass plates (silica gel 60 F254), and the compounds were visualized by dipping the plates in an ethanol solution of phosphomolybdic acid followed by heating (Merk & Co., Inc., Darmstadt, Germany). MW-assisted reactions were carried out using a Biotage Initiator® (Basel, Switzerland). Anhydrous CH2CH2 was purchased from Wako Pure Chemical Industries (Osaka, Japan).
4.1. Synthesis of (E)-Methyl 4-((1-Benzyl-1H-pyrazol-4-yl)oxy)but-2-enoate (1e)
To 1-benzyl-4-formyl-1H-pyrazole (200 mg, 1.07 mmol) in CH2Cl2 (5 mL) was added 70% meta-chloroperoxybenzoic acid (397.6 mg, 1.61 mmol) at 0 °C. After it was stirred overnight at room temperature, the mixture was quenched by adding aqueous NaHCO3 and then extracted with CH2Cl2. The organic layer was dried over MgSO4, filtered, and evaporated to give a crude residue. The crude material was dissolved in t-BuOH-CH2Cl2 (5 mL/5 mL) at 40 °C, and then potassium tert-butoxide (428.6 mg, 3.82 mmol) was added to the solution. After it was stirred overnight at 40 °C, the mixture was quenched with saturated aqueous NH4Cl and extracted with CH2Cl2. The separated organic layer was dried over MgSO4, filtered, and evaporated under reduced pressure to afford a crude residue, which was purified using silica gel column chromatography (eluent: EtOAc:hexane = 1:3) to afford (E/Z)-1e (128.1 mg, 44%): oil; IR (film) vmax 1724 (C=O), 1574 (C=C), 1437 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 3.74 (3H, s, -COOMe), 4.52 (2H, dd, J = 4.1, 1.9 Hz, -OCH2CH=CH-), 6.13 (1H, br d, J = 15.9 Hz, -COCH=CH-), 6.99 (1H, dt, J = 15.9, 4.1 Hz, -CH2CH=CH-), 7.05 (1H, d, J = 0.6 Hz, pyrazole-H), 7.18 (2H, br d, J = 8.0 Hz, Bn-H), 7.30–7.35 (4H, m, Bn-H, pyrazole-H); 13C NMR (100 MHz, CDCl3): δ 51.6, 56.6, 70.0, 115.1, 121.4, 127.2, 127.5, 128.0, 128.7, 136.3, 142.5, 145.2, 166.4; high-resolution electron ionization mass spectrometry (HREIMS) m/z calcd. for C15H16N2O3 (M+) 272.1161, found 272.1163.
*(E)-Methyl 4-((1-trityl-1H-pyrazol-4-yl)oxy)but-2-enoate (1f) was synthesized in a similar manner as 1e, but it was not rearranged under the thermal condition described below. 1f: colorless crystals (CH2Cl2); mp 155–158 °C; IR (film) vmax 1725 (C=O), 1572 (C=C), 1492 (C=C), 1442 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 3.76 (3H, s, -COOMe), 4.53 (2H, dd, J = 4.1, 2.0 Hz, -OCH2CH=CH-), 5.20 (2H, s, ArCH2Ph), 6.14 (1H, dt, J = 15.9, 1.9 Hz, -COCH=CH-), 7.00 (1H, dt, J = 15.9, 4.1 Hz, -CH2CH=CH-), 7.05 (1H, s, pyrazole-H), 7.13–7.18 (6H, m, Tr-H), 7.30–7.35 (9H, m, Tr-H), 7.42 (1H, s, pyrazole-H); 13C NMR (100 MHz, CDCl3): δ 51.7, 70.0, 78.7, 118.4, 121.5, 127.68, 127.71, 127.9, 130.1, 142.5, 143.0, 143.8, 166.4; HREIMS m/z calcd. for C27H24N2O3 (M+) 424.1786, found 424.1779.
4.2. Synthesis of Methyl 2-(1-Benzyl-4-hydroxy-1H-pyrazol-5-yl)but-3-enoate (2e)
A sealed microwave vial containing a solution of 1e (128.1 mg, 0.47 mmol) in 1,2-dimethoxyethane (DME) (2 mL) was heated under microwave irradiation at 200 °C for 30 min. After it had cooled, the reaction mixture was quenched with saturated aqueous NH4Cl and extracted with CH2Cl2. The separated organic layer was dried over MgSO4, filtered, and evaporated under reduced pressure to afford a crude residue, which was purified using silica gel column chromatography (eluent: EtOAc:hexane = 1:1) to afford 2e with a small amount of the isomer, 5e (53.9 mg, 42%).
2e (major) and 5e (minor) in ca. 2:1 ratio: oil; IR (film) vmax 1716 (C=O), 1497 (C=C), 1435 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 1.65 (1H, d, J = 7.3 Hz, =CHCH3 of 5e), 3.64 (1H, s, -COOMe of 5e), 3.68 (2H, s, -COOMe of 2e), 4.37 (0.7H, br d, J = 7.0 Hz, ArCH(COOMe)CH=), 4.93 (0.7H, dd, J = 17.0, 1.5 Hz, -CH=CHH), 5.01 (0.6H, s, ArCH2Ph), 5.12 (0.7H, dd, J = 10.3, 1.5 Hz, -CH=CHH of 2e), 5.19 (0.7H, br d, J = 16.1 Hz, ArCHHPh of 2e), 5.25 (0.7H, br d, J = 16.1 Hz, ArCHHPh of 2e), 5.86 (1H, ddd, J = 17.0, 10.3, 6.5 Hz, -CH(COOMe)CH=CH2 of 2e), 6.83 (0.6H, br s, -OH of 2e), 7.03–7.05 (2H, m, Ph-H), 7.19–7.31 (3H, m, Ph-H; 0.3H, m, overlapped, =CHCH3 of 5e), 7.30 (1H, br s, pyrazole-H); 13C NMR (150 MHz, CDCl3): δ 15.8 (5e), 46.4, 52.3 (5e), 53.2, 54.4 (5e), 54.6, 118.7, 120.6, 122.4 (5e), 122.9 (5e), 126.6, 127.2 (5e), 127.6 (5e), 127.9, 128.2 (5e), 128.40 (5e), 128.44, 129.1, 130.7, 136.9 (5e), 139.8 (5e), 140.9, 147.0, 166.5 (5e), 173.2; HREIMS m/z calcd. for C15H16N2O3 (M+) 272.1161, found 272.1162.
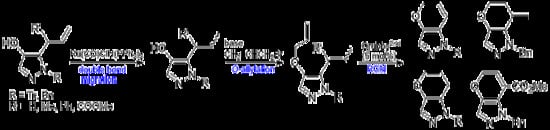
4.3. Double Bond Migration of 5-Allyl-4-hydroxy-1H-pyrazoles (Scheme 1)
General procedure: To a toluene solution (10 mL) of 5-allyl-4-hydroxy-1-trityl-1H-pyrazole (2a) (0.434 g, 1.19 mmol) in a microwave vial (5–20 mL), RuClH(CO)(PPh3)3 (56.6 mg, 0.059 mmol) was added. The reaction vial was sealed and then heated at 150 °C for 15 min under microwave irradiation. The cooled reaction mixture was evaporated to give a crude residue, which was purified using column chromatography (eluent: hexane:EtOAc = 1:1) to afford 4-hydroxy-5-(1-propenyl)-1-trityl-1H-pyrazole (5a) (0.323 g, 74% yield) as an E/Z mixture.
**Pure starting material gave the desired product as described above, but a small contamination inhibited the isomerization. In that case, a toluene-MeOH (9:1) solvent system was effective for isolating the desired product.
5a: oil; IR (film) vmax 3268 (-OH), 1597 (C=C), 1494 (C=C), 1446 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 1.43 (3H, dd, J = 6.5, 1.2 Hz, =CHCH3), 5.17 (1H, dd, J = 11.4, 1.4 Hz, ArCH=CH-), 5.23 (1H, dq, J = 11.4, 6.6 Hz, -CH=CHCH3), 7.09–7.34 (16H, m, Tr-H, pyrazole-H); 13C NMR (100 MHz, CDCl3): δ 14.9, 78.8, 118.3, 126.2, 127.3, 127.4, 127.6, 127.8, 129.6, 130.0, 130.1, 130.28, 130.34, 142.6; HREIMS m/z calcd. for C25H22N2O (M+) 366.1732, found 366.1731.
(E/Z)-1-Benzyl-4-hydroxy-5-(1-propenyl)-1H-pyrazole (5b): E/Z mixture in ca. 5:1 ratio (X); oil; IR (film) vmax 3031 (-OH), 1589 (C=C), 1496 (C=C), 1454 (C=C) cm−1; 1H NMR (600 MHz, CDCl3): δ 1.72 (0.5H, dd, J = 6.8, 1.5 Hz, -CH=CHCH3 of (E)-isomer), 1.83 (2.5H, dd, J = 6.8, 1.8 Hz, -CH=CHCH3 of (Z)-isomer), 5.15 (0.3H, s, -NCH2Ph of (Z)-isomer), 5.24 (1.5H, s, -NCH2Ph of (E)-isomer), 5.93 (0.15H, dq, J = 10.1, 6.8 Hz, -CH=CHCH3 of (Z)-isomer), 5.99 (0.15H, dq, J = 10.1, 1.5 Hz, ArCH=CHCH3 of (Z)-isomer), 6.15 (0.85H, dq, J = 16.1, 1.5 Hz, ArCH=CHCH3 of (E)-isomer), 6.38 (0.85H, dq, J = 16.1, 6.8 Hz, -CH=CHCH3 of (E)-isomer), 7.06–7.09 (6H, m, Tr-H), 7.16 (1H, s, pyrazole-H), 7.22–7.31 (9H, m, Tr-H); 13C NMR of (E)-isomer (150 MHz, CDCl3): δ 19.2, 53.7, 116.7, 126.6, 127.6, 127.9, 128.7, 130.3, 137.1, 138.9 (two carbon signals were deduced to have overlapped); (Z)-isomer: δ 15.4, 54.0, 115.4, 126.8, 126.9, 127.9, 128.6, 133.4, 137.0, 138.4 (two carbon signals were deduced to have overlapped); HREIMS m/z calcd. for C13H14N2O (M+) 214.1106, found 214.1104.
(E/Z)-1-Benzyl-4-hydroxy-5-(1-(1-methyl)propen-1-yl)-1H-pyrazole (5c): E/Z ratio = ca. 1:1; oil; IR (film) vmax 3063 (OH), 1563 (C=C), 1497 (C=C), 1456 (C=C) cm−1; 1H NMR of E/Z mixture (400 MHz, CDCl3): δ 1.41 (1.6H, dd, J = 6.9, 1.5 Hz, -CH=CHCH3), 1.73 (1.4H, dd, J = 6.8, 1.2 Hz, -CH=CHCH3), 1.79 (3H, br s, CqCH3), 4.30 (0.47H, br s, -OH), 4.43 (0.53H, br s, -OH), 5.08 (0.9H, s, -NCH2Ph), 5.15 (1.1H, s, -NCH2Ph), 5.57 (0.47H, qq, J = 6.8, 1.6 Hz, -CCH=CHCH3), 5.78 (0.53H, qq, J = 6.9, 1.6 Hz, -CCH3=CHCH3), 7.03 (0.94H, br d, J = 6.7 Hz, Ph-H), 7.07 (1.06H, br d, J = 6.7 Hz, Ph-H), 7.06–7.09 (6H, m, Ph-H), 7.16–7.36 (3H, m, Ph-H), 7.21 (1H, s, pyrazole-H); 13C NMR of E/Z mixture (100 MHz, CDCl3): δ 14.0, 15.0, 16.2, 32.0, 53.9, 54.1, 124.2, 124.6, 126.8, 127.2, 127.4, 127.5, 127.91, 127.94, 128.46, 128.49, 128.6, 129.8, 130.0, 132.6, 137.3, 137.6, 137.8; HREIMS m/z calcd. for C14H16N2O (M+) 228.1263, found 228.1260.
(E/Z)-1-Benzyl-4-hydroxy-5-(1-(1-phenyl)propenyl)-1H-pyrazole (5d): isomer ratio = ca. 5:1; oil; IR (film) vmax 3031 (OH), 1573 (C=C), 1496 (C=C) cm−1; 1H NMR (100 MHz, CDCl3): δ 1.55 (2.5H, d, J = 7.0 Hz, =CHCH3), 1.85 (0.5H, d, J = 7.3 Hz, =CHCH3), 4.61 (1H, br s, J = 14.8 Hz, -NCHHPh), 4.95 (1H, br s, J = 15.3 Hz, -NCHHPh), 5.94 (0.16H, q, J = 7.2 Hz, Cq=CHCH3), 6.31 (0.84H, br q, J = 7.0 Hz, Cq=CHCH3), 6.87–6.90 (0.66H, m, Ph-H), 6.93–6.96 (1.34H, m, Ph-H), 7.06–7.34 (4H, m, Ph-H), 7.32 (1H, s, pyrazole-H); 13C NMR (100 MHz, CDCl3): δ 15.7, 54.3, 126.2, 127.0, 127.38, 127.45, 127.6, 128.3, 128.4, 128.6, 128.9, 129.3, 131.1, 136.9, 139.9 (minor isomer: 15.4, 54.0, 126.4, 127.68, 127.8); HREIMS m/z calcd. for C19H18N2O (M+) 290.1419, found 290.1417.
(E/Z)-1-Benzyl-4-hydroxy-5-(1-(1-methoxycarbonyl)propenyl)-1H-pyrazole (5e): E/Z mixture in ca. 13:1 ratio; oil; IR (film) vmax 3090 (OH), 1716 (C=O), 1507 (C=C), 1436 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 1.72 (2.6H, d, J = 7.3 Hz, -CH=CHCH3 of major isomer), 1.83 (0.4H, d, J = 7.2 Hz, -CH=CHCH3 of minor isomer), 3.66 (2.6H, s, -OCH3 of major isomer), 3.71 (0.4H, s, -OCH3 of minor isomer), 4.73 (1H, s, -OH), 5.06 (1.86H, s, -NCH2Ph of major isomer), 5.17 (0.14H, s, -NCH2Ph of minor isomer), 7.04 (2H, br d, J = 6.6 Hz, Ph-H), 7.19–7.30 (4H, m, Ph-H, =CHCH3), 7.33 (1H, s, pyrazole-H); 13C NMR (100 MHz, CDCl3): δ 15.8, 52.3, 54.5, 122.2, 122.9, 127.3, 127.6, 128.1, 128.4, 136.7, 140.0, 147.5, 166.5; HREIMS m/z calcd. for C15H16N2O3 (M+) 272.1161, found 272.1160.
4.4. O-Allylation of 1-Protected 5- or 3-Allyl-4-allyloxy-1H-pyrazoles (Scheme 1)
General procedure: To a solution of an E/Z mixture of 4-hydroxy-5-(1-propenyl)-1H-1-tritylpyrazole (5a) (0.410 g, 1.12 mmol) in acetone (2 mL), 20% aqueous NaOH (1 mL) and allyl bromide (142 μL, 1.68 mmol) were added. The reaction mixture was stirred for 1 h and then quenched with saturated aqueous NH4Cl and extracted with CH2Cl2. The organic layer was dried over anhydrous MgSO4, filtered, and evaporated. The crude residue was purified with column chromatography (eluent: hexane:EtOAc = 3:1) to afford 4-allyloxy-5-(1-propenyl)-1H-1-tritylpyrazole (6a) (E/Z mixture in ca. 3:1 ratio, 0.334 g, 73% yield).
(E)-6a: mp 152–155 °C; IR (KBr) vmax 1567 (C=C), 1491 (C=C), 1446 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 1.40 (3H, dd, J = 6.7, 1.5 Hz, CH3CH=), 4.50 (2H, dt, J = 5.3, 1.5 Hz, -OCH2CH=CH2), 5.26 (1H, dq, J = 15.8, 1.4 Hz, -CH2CH=CHH), 5.37 (1H, dq, J = 17.2, 1.5 Hz, -CH2CH=CHH), 5.46 (1H, dq, J = 15.8, 1.4 Hz, ArCH=CHCH3), 6.04 (1H, ddt, J = 17.2, 10.5, 5.3 Hz, -OCH2CH=CH2), 6.14 (1H, dq, J = 15.8, 6.7 Hz, ArCH=CHCH3), 7.07–7.16 (6H, m, Tr-H), 7.24–7.39 (9H, m, Tr-H), 7.32 (1H, s, pyrazole-H); 13C NMR (100 MHz, CDCl3): δ 18.9, 72.0, 79.0, 117.5, 120.0, 124.7, 127.3, 127.4, 127.6, 128.5, 130.3, 133.5, 142.9, 143.5; HREIMS m/z calcd. for C28H26N2O (M+) 406.2055, found 406.2047.
(Z)-6a: mp 82–86 °C; IR (KBr) vmax 1567 (C=C), 1491 (C=C), 1446 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 1.42 (3H, d, J = 5.1 Hz, CH3CH=), 4.47 (2H, dt, J = 5.5, 1.6 Hz, -OCH2CH=CH2), 5.16–5.20 (2H, m), 5.22 (1H, dq, J = 10.6, 1.4 Hz, -CH2CH=CHH), 5.34 (1H, dq, J = 17.2, 1.5 Hz, -CH2CH=CHH), 6.00 (1H, ddt, J = 17.2, 10.6, 5.6 Hz, -OCH2CH=CH2), 7.03–7.17 (6H, m, Tr-H), 7.21–7.32 (9H, m, Tr-H), 7.37 (1H, s, pyrazole-H); 13C NMR (100 MHz, CDCl3): δ 15.4, 72.0, 78.9, 117.4, 117.9, 124.9, 127.2, 127.3, 127.9, 129.8, 130.1, 133.7, 142.6, 142.9; HREIMS m/z calcd. for C28H26N2O (M+) 406.2046, found 406.2050.
(E/Z)-4-Allyloxy-1-benzyl-5-(1-propenyl)-1H-pyrazole (6b) (an inseparable E/Z mixture in a ca. 8:2 ratio, 0.334 g, 73% yield): oil; IR (film) vmax 1566 (C=C), 1495 (C=C), 1452 (C=C) cm−1; HREIMS m/z calcd. for C16H18N2O (M+) 254.1419, found 254.1421. (E)-isomer: 1H NMR (600 MHz, CDCl3): δ 1.82 (3H, d, J = 6.7, 1.8 Hz, CH3CH=), 4.50 (2H, dt, J = 5.4, 1.4 Hz, -OCH2CH=CH2), 5.26 (1H, dq, J = 10.6, 1.4 Hz, -CH2CH=CHH), 5.28 (2H, s, NCH2Ph), 5.39 (1H, ddd, J = 17.3, 3.2, 1.8 Hz, -CH2CH=CHH), 6.05 (1H, ddt, J = 17.3, 10.6, 5.4 Hz, -OCH2CH=CH2), 6.16 (1H, br d, J = 15.8 Hz, ArCH=CHCH3), 6.46 (1H, dq, J = 15.8, 6.7 Hz, -CH=CHCH3), 7.07 (2H, br d, J = 7.6 Hz, Ph-H), 7.24 (1H, br t, J = 7.6 Hz, Ph-H), 7.26 (1H, s, pyrazole-H), 7.30 (2H, br t, J = 7.6 Hz, Ph-H); 13C NMR (150 MHz, CDCl3): δ 19.3, 53.9, 72.2, 116.6, 117.5, 125.8, 126.5, 126.8, 127.5, 128.6, 128.7, 129.6, 133.5, 137.2; (Z)-isomer: 1H NMR (600 MHz, CDCl3): δ 1.72 (3H, dd, J = 6.8, 1.8 Hz, CH3CH=), 4.46 (2H, dt, J = 5.6, 1.5 Hz, -OCH2CH=CH2), 5.18 (2H, s, NCH2Ph), 5.24 (1H, dq, J = 10.5, 1.5 Hz, -CH2CH=CHH), 5.36 (1H, dq, J = 17.1, 1.5 Hz, -CH2CH=CHH), 5.91 (1H, dq, J = 11.2, 6.8 Hz, -CH=CHCH3), 6.01 (1H, ddt, J = 17.1, 10.5, 5.6 Hz, -OCH2CH=CH2), 6.01 (1H, br d, J = 11.2 Hz, ArCH=CHCH3, overlapped), 7.07 (2H, br d, J = 7.6 Hz, Ph-H), 7.24 (1H, br t, J = 7.6 Hz, Ph-H), 7.28 (1H, s, pyrazole-H), 7.30 (2H, br t, J = 7.6 Hz, Ph-H); 13C NMR (150 MHz, CDCl3): δ 15.7, 53.9, 72.4, 115.4, 117.4, 126.59, 126.64, 127.5, 133.2, 137.1, 142.7 (three signals should be overlapped with signals of the (E)-isomer).
(E/Z)-4-Allyloxy-1-benzyl-5-(1-(1-methyl)propenyl)-1H-pyrazole (6c): isomer ratio = ca. 1:1; oil; IR (film) vmax 1562 (C=C), 1496 (C=C), 1455 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 1.43 (1.6H, dd, J = 6.7, 1.4 Hz, CH3CH=), 1.72 (1.4H, dd, J = 6.9, 1.2 Hz, CH3CH=), 4.414 (1.06H, d, J = 5.5 Hz, -OCH2CH=), 4.417 (0.94H, d, J = 5.5 Hz, -OCH2CH=), 5.08 (0.94H, s, NCH2Ph), 5.19 (1.06H, s, NCH2Ph), 5.19–5.36 (2H, m, =CH2), 5.54 (0.44H, qq, J = 6.9, 1.6 Hz, -C(CH3)H=CH3), 5.75 (0.56H, qq, J = 6.8, 1.6 Hz, -C(CH3)H=CH3), 5.92–6.04 (1H, m, -CH2CH=CH2), 7.02 (0.94H, br d, J = 7.3 Hz, Ph-H), 7.07 (1.06H, br d, J = 7.2 Hz, Ph-H), 7.18–7.29 (3H, m, Ph-H), 7.29 (1H, s, pyrazole-H); 13C NMR (100 MHz, CDCl3): δ 13.9, 15.2, 16.0, 23.1, 53.7, 54.0, 72.8, 117.45, 117.54, 125.0, 126.77, 126.86, 127.21, 127.3, 127.5, 128.46, 128.49, 129.0, 129.2, 129.5, 133.2, 133.7, 133.8, 137.3, 137.8, 141.6, 141.7; HREIMS m/z calcd. for C17H20N2O (M+) 268.1576, found 268.1575.
(E/Z)-4-Allyloxy-1-benzyl-5-(1-(1-phenyl)propenyl)-1H-pyrazole (6d): isomer ratio = ca. 7:1; oil; IR (film) vmax 1556 (C=C), 1500 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 1.55 (2.7H, d, J = 6.9 Hz, CH3CH=), 1.85 (0.3H, d, J = 7.3 Hz, CH3CH=), 4.38 (0.25H, br d, J = 5.5 Hz, -OCH2CH=), 4.44 (1.75H, br d, J = 3.9 Hz, -OCH2CH=CH2), 4.64 (1H, br d, J = 14.9 Hz, NCHHPh), 4.94 (1H, br d, J = 14.8 Hz, NCHHPh), 5.19 (1H, dd, J = 10.5, 1.3 Hz, -CH2CH=CHH), 5.29 (1H, dq, J = 17.2, 1.6 Hz, -CH2CH=CHH), 5.89–6.99 (1H, m, -OCH2CH=CH2 overlaps with 0.12H, m, =CHCH3), 6.31 (0.88H, q, J = 7.0 Hz, =CHCH3), 6.89–6.90 (2H, m, Ph-H), 7.08–7.49 (8H, m, Ph-H), 7.39 (1H, s, pyrazole-H); 13C NMR (100 MHz, CDCl3): δ 15.8, 54.3, 72.7, 117.5, 126.2, 127.0, 127.1, 127.4, 128.27, 128.33, 128.5, 129.2, 129.3, 131.2, 133.7, 137.0, 139.7, 143.3; HREIMS m/z calcd. for C22H22N2O (M+) 330.1732, found 330.1729.
Synthesis of (E/Z)-4-allyloxy-1-benzyl-5-(1-(1-methoxycarbonyl)propenyl)-1H-pyrazole (6e) from 2e: To an acetone solution (4.5 mL) of 2e with a small amount of 5e (121.8 mg, 0.45 mmol) in a microwave vial were added K2CO3 (61.8 mg, 0.45 mmol) in water (0.5 mL) and allyl bromide (0.04 mL, 0.45 mmol). After the reaction vial was sealed, the mixture was heated under microwave irradiation at 60 °C for 1 h. After it had cooled, the reaction was quenched by adding aqueous NH4Cl. Then, the reaction mixture was extracted with EtOAc three times. The organic layer was washed with brine, dried over MgSO4, filtered, and then evaporated to give a crude residue, which was purified using column chromatography (eluent: hexane:EtOAc = 2:1) to give pure 6e (117.1 mg, 84%).
6e (isomer ratio = ca. 13:1): oil; IR (film) vmax 1717 (C=O), 1500 (C=C) cm−1; 1H NMR: δ 1.54 (2.6H, d, J = 7.2 Hz, CH3CH= of major isomer), 2.12 (0.4H, d, J = 7.2 Hz, CH3CH= of minor isomer), 3.52 (0.4H, s, -OCH3 of minor isomer), 3.60 (2.6H, s, -OCH3 of major isomer), 4.92 (0.93H, br d, J = 15.3 Hz, NCHHPh of major isomer), 5.01 (0.14H, s, NCH2Ph of minor isomer), 5.03 (0.93H, br d, J = 15.3 Hz, NCHHPh of major isomer), 5.11 (0.93H, dq, J = 10.6, 1.4 Hz, -CH2CH=CHH of major isomer), 5.14 (0.07H, dq, J = 10.6, 1.5 Hz, -CH2CH=CHH of minor isomer), 5.22 (0.93H, dq, J = 17.5, 1.6 Hz, -CH2CH=CHH of major isomer), 5.23 (1H, dq, J = 17.4, 1.6 Hz, -CH2CH=CHH of minor isomer), 5.87–5.93 (1H, m, -OCH2CH=CH2), 6.46 (0.07H, q, J = 7.3 Hz, -Cq=CHCH3 of minor isomer), 7.06 (2H, br d, J = 6.6 Hz, Ph-H), 7.17–7.30 (3H, m, Ph-H), 7.20 (0.07H, q, J = 7.2 Hz, -Cq=CHCH3 of major isomer), 7.32 (1H, s, pyrazole-H); 13C NMR (100 MHz, CDCl3): δ 15.7, 52.0, 54.6, 72.5, 117.6, 122.0, 123.2, 126.4, 127.4, 127.6, 128.4, 133.4, 136.7, 143.2, 147.9, 165.9 (minor isomer: 16.2, 51.5, 54.1, 72.8, 121.5, 123.1, 126.6, 127.5, 136.9, 147.7); HREIMS m/z calcd. for C18H20N2O3 (M+) 312.1474, found 312.1467.
4.5. Ring-Closing Metathesis of 6 to 1H-1,5-Dihydropyrano[3,2-c]pyrazoles 7 (Table 1)
General procedure (
Table 1, entry 3): To a solution of
6a (21.8 mg, 0.054 mmol) in CH
2Cl
2 (2 mL) was added Grubbs
2nd (1.7 mg, 2.7 mmol) at rt. The reaction mixture was stirred at rt for 1 h, and then the solvent was removed under reduced pressure, affording a crude residue, which was purified using silica gel column chromatography (eluent: EtOAc:hexane = 1:3) to afford
7a (16.2 mg, 83%).
*General procedure for MW-aided reaction (
Table 1, entry 5): To a solution of
6a (16.4 mg, 0.04 mmol) in CH
2Cl
2 (2 mL) was added Grubbs
2nd (2.3 mg, 2.0 mmol) in a microwave vial. The reaction mixture was heated under microwave irradiation at 80 °C for 3 min. After the reaction mixture had cooled, the solvent was removed under reduced pressure, affording a crude residue, which was purified using silica gel column chromatography (eluent: EtOAc:hexane = 1:4) to afford
7a (12.8 mg, 87%).
1,5-Dihydro-1-tritylpyrano[3,2-c]pyrazole (7a): oil; IR (film) vmax 1677 (C=C), 1581 (C=C), 1493 (C=C), 1447 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 4.64 (2H, dd, J = 3.8, 1.8 Hz, -OCH2CH=CH-), 5.15 (1H, dt, J = 10.2, 3.7 Hz, -OCH2CH=CH-), 5.28 (1H, dtd, J = 10.2, 1.8, 0.8 Hz, -OCH2CH=CH-), 7.08–7.17 (6H, m, Tr-H), 7.18 (1H, d, J = 0.8 Hz, pyrazole-H), 7.23–7.32 (9H, m, Tr-H); 13C NMR (100 MHz, CDCl3): δ 66.9, 78.0, 117.7, 118.6, 124.3, 127.55, 127.57, 130.1, 141.4, 142.7; HREIMS m/z calcd. for C25H20N2O (M+) 364.1575, found 364.1585.
1-Benzyl-1,5-dihydropyrano[3,2-c]pyrazole (7b): oil; IR (film) vmax 1566 (C=C), 1495 (C=C), 1452 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 4.75 (2H, dd, J = 3.9, 1.8 Hz, -OCH2CH=), 5.21 (2H, s, ArCH2Ph), 5.53 (1H, dt, J = 10.0, 3.9 Hz, -OCH2CH=CH-), 6.34 (1H, br d, J = 10.0 Hz, -OCH2CH=CH-), 7.10 (1H, d, J = 0.8 Hz, pyrazole-H), 7.10–7.14 (2H, d, J = 6.6 Hz, Ph-H), 7.26–7.32 (3H, m, Ph-H); 13C NMR (100 MHz, CDCl3): δ 54.0, 67.2, 115.5, 119.7, 124.5, 127.1, 127.9, 128.8, 136.6, 140.9; HREIMS m/z calcd. for C13H12N2O (M+) 212.0950, found 212.0949.
1-Benzyl-1,5-dihydro-7-methylpyrano[3,2-c]pyrazole (7c): oil; IR (film) vmax 1732 (C=O), 1541 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 1.96 (3H, br s, CqCH3), 4.64 (2H, dq, J = 3.3, 1.6 Hz, -OCH2CH=), 5.23–5.26 (1H, m, -OCH2CH=), 5.39 (2H, s, NCH2Ph), 7.01 (2H, br d, J = 7.0 Hz, Ph-H), 7.17 (1H, s, pyrazole-H), 7.24–7.32 (3H, m, Ph-H); 13C NMR (100 MHz, CDCl3): δ 18.1, 55.3, 67.6, 116.5, 124.7, 126.0, 127.6, 127.3, 128.7, 137.7, 141.4; HREIMS m/z calcd. for C15H14N2O3 (M+) 270.1004, found 270.1003.
1-Benzyl-1,5-dihydro-7-methoxycarbonylpyrano[3,2-c]pyrazole (7e): oil; IR (film) vmax 1732 (C=O), 1541 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 3.76 (3H, s, -COOCH3), 4.72 (2H, d, J = 4.5 Hz, -OCH2CH=), 5.57 (2H, s, ArCH2Ph), 6.45 (1H, t, J = 4.5 Hz, -OCH2CH=Cq), 6.33 (1H, br d, J = 10.0 Hz, -OCH2CH=CH-), 7.04 (2H, br d, J = 6.5 Hz, Ph-H), 7.23 (1H, s, pyrazole-H), 7.23–7.31 (3H, m, Ph-H); 13C NMR (100 MHz, CDCl3): δ 52.3, 56.4, 66.9, 124.0, 124.7, 127.0, 127.4, 128.4, 128.7, 137.5, 142.4, 163.8; HREIMS m/z calcd. for C15H14N2O3 (M+) 270.1004, found 270.1003.
1-Benzyl-1,7-dihydropyrano[3,2-c]pyrazole (8b): oil; IR (film) vmax 1607 (C=C), 1586 (C=C), 1557 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 3.23 (2H, dd, J = 3.3, 2.0 Hz, ArCH2CH=), 4.77 (1H, dt, J = 6.3, 3.4 Hz, -CH2CH=CH-), 5.17 (2H, s, ArCH2Ph), 6.42 (1H, dt, J = 6.2, 2.0 Hz, =CH=CHO-), 7.06–7.20 (2H, m, Ph-H), 7.22–7.33 (4H, m, Ph-H, pyrazole-H); 13C NMR (100 MHz, CDCl3): δ 19.5, 53.8, 97.1, 125.1, 126.4, 127.0, 127.9, 128.8, 129.0, 136.6, 141.3; HREIMS m/z calcd. for C13H12N2O (M+) 212.0950, found 212.0947.
1-Benzyl-1,7-dihydro-7-methylenepyrano[3,2-c]pyrazole (9c): oil; IR (film) vmax 1644 (C=C), 1556 (C=C), 1401 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 2.54 (2H, br t, J = 5.6 Hz, -OCH2CH2Cq), 4.17 (2H, t, J = 5.7 Hz, -OCH2CH2-), 4.78 (1H, br s, CqCHH), 4.96 (1H, br s, CqCHH), 5.43 (2H, s, NCH2Ph), 7.02 (2H, d, J = 7.0 Hz, Ph-H), 7.22–7.32 (4H, m, Ph-H, pyrazole-H); 13C NMR (100 MHz, CDCl3): δ 32.2, 55.4, 68.3, 107.2, 124.0, 125.3, 126.2, 127.5, 128.7, 129.7, 136.9, 142.6; HREIMS m/z calcd. for C14H14N2O (M+) 226.1106, found 226.1102.
1,4-Bis((1-benzyl-5-(1-phenylprop-1-en-1-yl)-1H-pyrazol-4-yl)oxy)but-2-ene (10d): oil; IR (film) vmax 1569 (C=C), 1496 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 1.51 (6H, d, J = 7.1 Hz, =CHCH3), 4.40 (4H, br s, -OCH2CH=), 4.62 (2H, br d, J = 14.8 Hz, ArCHHPh), 4.92 (2H, br d, J = 14.4 Hz, ArCHHPh), 5.82–5.84 (2H, m, -OCH2CH=), 6.29 (2H, q, J = 7.1 Hz, =CHCH3), 6.92–6.95 (4H, m, Ph-H), 7.06–7.25 (6H, m, Ph-H), 7.35 (2H, s, pyrazole-H); 13C NMR (100 MHz, CDCl3): δ 15.8, 54.3, 71.6, 126.1, 127.1, 127.35, 127.42, 128.3, 128.7, 129.1, 129.2, 131.2, 137.0, 140.0, 143.2 (three carbon signals overlapped); HREIMS m/z calcd. for C42H40N4O2 (M+) 632.3151, found 632.3145.
1,4-Bis((1-benzyl-5-(1-(methoxycarbonyl)prop-1-en-1-yl)-1H-pyrazol-4-yl)oxy)but-2-ene (10e): oil; IR (film) vmax 1722 (C=O), 1712 (C=O), 1642 (C=C), 1573 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 1.54 (5.4H, d, J = 7.0 Hz, =CHCH3 of major isomer), 2.14 (0.6H, d, J = 7.2 Hz, =CHCH3 of minor isomer), 3.61 (0.6H, s, -OCH3 of minor isomer), 3.62 (5.4H, s, =CHCH3 of major isomer), 4.42 (3.6H, br s, -OCH2CH= of major isomer), 4.48 (0.4H, br s, -OCH2CH= of minor isomer), 5.08 (1.8H, br d, J = 13.3 Hz, ArCHHPh of major isomer), 5.11 (0.4H, s, ArCH2Ph of minor isomer), 5.12 (1.8H, br d, J = 13.3 Hz, ArCHHPh of major isomer), 5.77 (3.6H, br t, J = 3.7 Hz, -OCH2CH= of minor isomer), 5.88 (0.4H, br t, J = 3.7 Hz, -OCH2CH= of major isomer), 6.28 (0.2H, q, J = 7.5 Hz, =CHCH3 of minor isomer), 7.08 (4H, d, J = 6.8 Hz, Ph-H), 7.20–7.32 (7.8H, m, Ph-H, =CHCH3 of major isomer), 7.33 (2H, s, pyrazole-H); 13C NMR (100 MHz, CDCl3): δ 15.7, 52.1, 54.6, 71.5, 122.1, 123.2, 126.4, 127.4, 127.6, 128.4, 128.5, 136.7, 147.9, 165.9; HREIMS m/z calcd. for C34H36N4O6 (M+) 596.2635, found 596.2634.
Methyl 2-(1-benzyl-4-(cinnamyloxy)-1H-pyrazol-5-yl)but-2-enoate (11e): oil; IR (film) vmax 1716 (C=O), 1644 (C=C), 1574 (C=C) cm−1; 1H NMR (600 MHz, CDCl3): δ 1.58 (3H, d, J = 7.3 Hz, =CHCH3), 3.58 (3H, s, -COOCH3), 4.58 (2H, d, J = 6.2 Hz, -OCH2CH=), 5.02 (1H, br d, J = 15.2 Hz, ArCHHPh), 5.12 (1H, br d, J = 15.2 Hz, ArCHHPh), 6.30 (1H, dt, J = 15.9, 6.2 Hz, -OCH2CH=CH-), 6.62 (1H, d, J = 15.9 Hz, -CH=CHPh), 7.09 (2H, d, J = 7.3 Hz, Ph-H), 7.20–7.38 (8H, m, Ph-H), 7.30 (1H, q, J = 7.3 Hz, -Cq=CHCH3), 7.40 (1H, s, pyrazole-H); 13C NMR (150 MHz, CDCl3): δ 15.8, 52.0, 54.7, 72.7, 122.2, 123.6, 124.7, 126.6, 126.9, 127.4, 127.6, 127.9, 128.4, 128.6, 133.1, 136.4, 136.7, 143.2, 147.9, 165.9; HREIMS m/z calcd. for C24H24N2O3 (M+) 388.1787, found 388.1785.
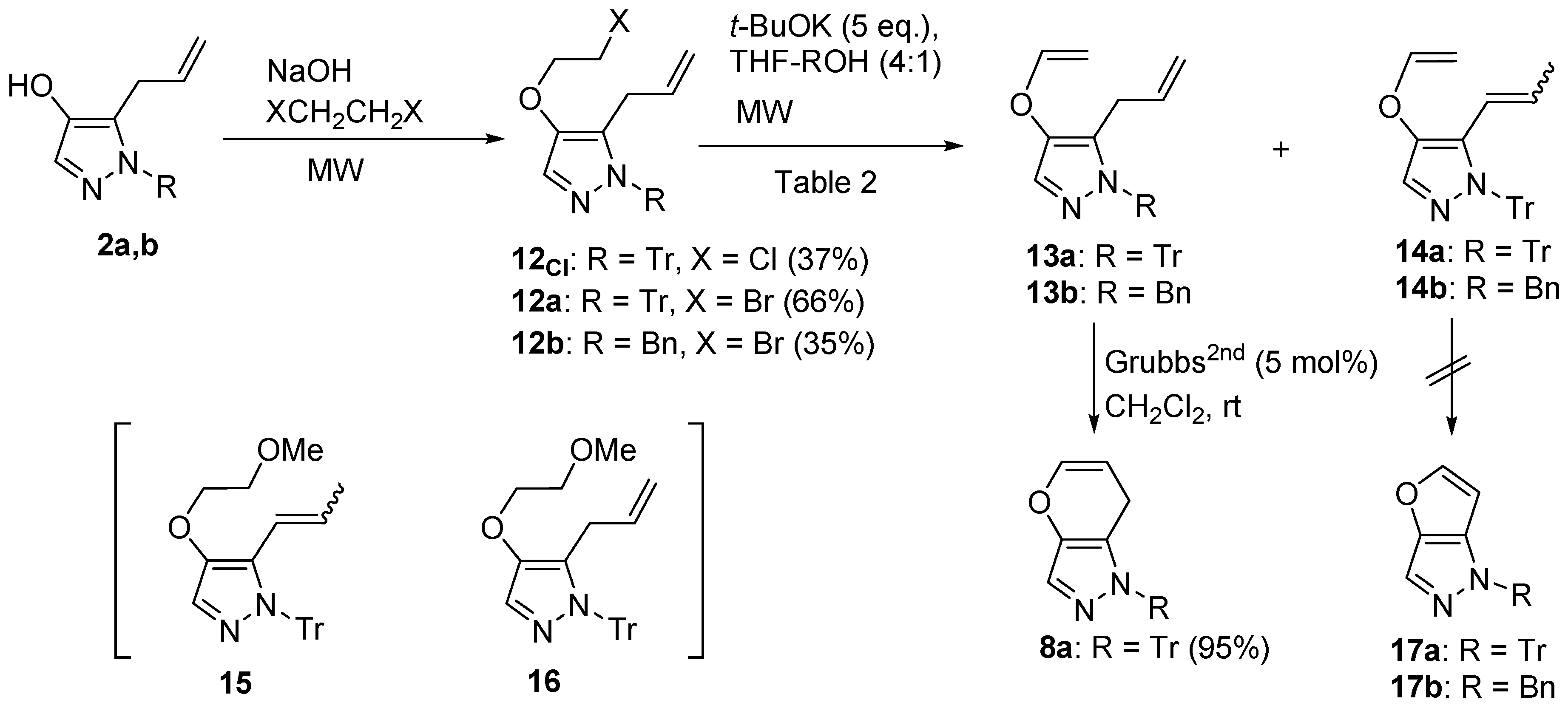
4.6. Synthesis of 5-Allyl-4-(2-haloethoxy)-1H-pyrazoles (12) (Scheme 2)
General procedure: To a solution of 2a (50.8 mg, 0.14 mmol) in acetone (2 mL) in a microwave vial were added 1,2-dibromoethane (0.05 mL, 0.56 mmol), 20% aqueous NaOH (0.11 mL, 0.56 mmol), and a catalytic amount of tetrabutylammonium bromide. The sealed reaction vial was MW irradiated at 140 °C for 30 min. After it had cooled, the reaction mixture was quenched with saturated aqueous NH4Cl and extracted with CH2Cl2. The separated organic layer was dried over MgSO4, filtered, and evaporated under reduced pressure to afford a crude residue. The residue was purified using silica gel column chromatography (eluent: EtOAc:hexane = 1:3) to afford 12a (42.9 mg, 65%) as an oil.
5-Allyl-4-(2-bromoethoxy)-1H-1-tritylpyrazole (12a): pale yellow crystals (CH2Cl2); mp 135–140 °C; IR (film) vmax 1581 (C=C), 1491 (C=C), 1446 (C=C) cm−1; 1H NMR (500 MHz, CDCl3): δ 2.85 (2H, dt, J = 6.5, 1.2 Hz, ArCH2CH=CH2), 3.56 (2H, t, J = 6.2 Hz, -OCH2CH2CBr), 4.20 (2H, t, J = 6.2 Hz, -OCH2CH2Br), 4.63 (1H, dq, J = 17.0, 1.6 Hz, -CH=CHH), 4.66 (1H, dq, J = 10.0, 1.4 Hz, -CH=CHH), 4.97 (1H, ddt, J = 17.0, 10.0, 6.5 Hz, -CH2CH=CH2), 7.10–7.13 (6H, m, Tr-H), 7.25–7.30 (9H, m, Tr-H), 7.33 (1H, s, pyrazole-H); 13C NMR (125 MHz, CDCl3): δ 29.4, 31.2, 71.6, 78.7, 115.9, 125.6, 127.4, 127.6, 129.9, 130.1, 132.4, 142.8, 143.6; HREIMS m/z calcd. for C27H25BrN2O (M+) 472.1151, found 472.1149.
5-Allyl-1-benzyl-4-(2-bromoethoxy)-1H-pyrazole (12b): oil; IR (film) vmax 1583 (C=C), 1496 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 3.29 (2H, dd, J = 4.7, 1.7 Hz, ArCH2CH=), 3.56 (2H, br t, J = 6.2 Hz, -OCH2CH2Br), 4.20 (2H, br t, J = 6.2 Hz, -OCH2CH2Br), 5.00 (1H, dd, J = 7.0, 1.4 Hz, -CH=CHH), 5.07 (1H, dd, J = 10.2, 1.4 Hz, -CH=CHH), 5.73–5.83 (1H, m, -CH2CH=CH2), 7.06 (2H, br d, J = 8.1 Hz, Bn-H), 7.25–7.33 (4H, m, Ph-H, pyrazole-H); 13C NMR (100 MHz, CDCl3): δ 27.1, 29.5, 53.9, 72.4, 116.5, 126.7, 127.2, 127.7, 127.8, 128.7, 133.6, 136.9, 141.6; HREIMS m/z calcd. for C15H17BrN2O (M+) 320.0524, found 320.0520.
5-Allyl-4-(2-chloroethoxy)-1H-1-tritylpyrazole (12Cl): white powder (CH2Cl2); mp 120–125 °C; IR (KBr) vmax 1580 (C=C), 1493 (C=C), 1446 (C=C) cm−1; 1H NMR (500 MHz, CDCl3): δ 2.85 (2H, br d, J = 7.6 Hz, ArCH2CH=), 3.72 (2H, t, J = 5.7 Hz, -OCH2CH2Cl), 4.14 (2H, t, J = 5.7 Hz, -OCH2CH2CCl), 4.63 (1H, dq, J = 17.0, 1.6 Hz, -CH=CHH), 4.66 (1H, dq, J = 10.0, 1.4 Hz, -CH=CHH), 4.97 (1H, ddt, J = 17.0, 10.0, 6.7 Hz, -CH2CH=CH2), 7.10–7.14 (6H, m, Tr-H), 7.24–7.31 (9H, m, Tr-H), 7.34 (1H, s, pyrazole-H); 13C NMR (125 MHz, CDCl3): δ 31.2, 42.1, 71.7, 78.6, 115.8, 125.5, 127.3, 127.6, 129.9, 130.0, 132.4, 142.8, 143.7; HREIMS m/z calcd. for C27H25ClN2O (M+) 428.1655, found 428.1654. *MW conditions: 160 °C, 30 min.
4.7. Reaction of 12 with Potassium Tert-Butoxide (Table 2, Scheme 2)
General procedure (
Table 2, entry 7): To a solution of
12a (28.8 mg, 0.05 mmol) in anhydrous THF:
t-BuOH (2 mL:0.5 mL) in a microwave vial was added potassium
tert-butoxide (28.8 mg, 0.26 mmol). The sealed reaction vial was MW irradiated at 130 °C for 1 h. After it had cooled, the reaction mixture was quenched with saturated aqueous NH
4Cl and extracted with CH
2Cl
2. The separated organic layer was dried over MgSO
4, filtered, and evaporated under reduced pressure to afford a crude residue. The residue was purified using silica gel column chromatography (eluent: EtOAc:hexane = 1:3) to afford
13a (20.8 mg, 87%).
5-Allyl-1-trityl-1H-4-vinyloxypyrazole (13a): white powder (CH2Cl2); mp 75–80 °C; IR (KBr) vmax 1639 (C=C), 1624 (C=C), 1566 (C=C) cm−1; 1H NMR (600 MHz, CDCl3): δ 2.81 (2H, ddd, J = 6.8, 1.5, 1.2 Hz, ArCH2CH=CH2), 4.23 (1H, dd, J = 5.4, 1.8 Hz, -OCH=CHH), 4.50 (1H, dd, J = 13.8, 2.1 Hz, -OCH2=CHH), 4.62 (1H, dq, J = 16.7, 1.5 Hz, -CH2CH=CHH), 4.68 (1H, dq, J = 10.9, 1.5 Hz, -CH2CH=CHH), 4.99 (1H, ddt, J = 16.5, 10.9, 2.1 Hz, -CH2CH=CH2), 6.53 (1H, dd, J = 13.8, 6.5 Hz, -OCH=CH2), 7.12–7.14 (6H, m, Tr-H), 7.25–7.31 (9H, m, Tr-H), 7.40 (1H, s, pyrazole-H); 13C NMR (150 MHz, CDCl3): δ 31.1, 77.8, 91.3, 116.1, 127.4, 127.6, 128.3, 130.0, 131.7, 131.9, 140.4, 142.3, 150.7; HREIMS m/z calcd. for C27H24N2O (M+) 392.1888, found 392.1880.
5-(1-Propenyl)-1-trityl-1H-4-vinyloxypyrazole (14a): white powder (CH2Cl2); mp 133–135 °C; IR (KBr) vmax 1639 (C=C), 1560 (C=C), 1492 (C=C) cm−1; 1H NMR (600 MHz, CDCl3): δ 1.39 (3H, dd, J = 6.8, 1.8 Hz, -CH=CHCH3), 4.29 (1H, dd, J = 6.2, 2.0 Hz, -OCH=CHH), 4.59 (1H, dd, J = 13.8, 2.0 Hz, -OCH=CHH), 5.39 (1H, br dq, J = 15.8, 0.8 Hz, -CH=CHCH3), 5.98 (1H, dq, J = 15.8, 6.8 Hz, -CH=CHCH3), 6.56 (1H, dd, J = 13.8, 6.2 Hz, -OCH=CH2), 7.11–7.15 (6H, m, Tr-H), 7.26–7.32 (9H, m, Tr-H), 7.38 (1H, br s, pyrazole-H); 13C NMR (150 MHz, CDCl3): δ 18.8, 79.8, 92.3, 119.1, 127.38, 127.44, 128.0, 129.1, 130.3, 131.2, 139.5, 142.7, 150.4; HREIMS m/z calcd. for C27H24N2O (M+) 392.1889, found 392.1887.
(E/Z)-1-Benzyl-5-(1-propenyl)-1H-4-vinyloxypyrazole (14b): E/Z ratio = ca. 5:1; oil; IR (film) vmax 1642 (C=C), 1562 (C=C), 1493 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 1.68 (0.5H, d, J = 6.3 Hz, =CHCH3 of (Z)-isomer), 1.82 (2.5H, dd, J = 6.6, 1.6 Hz, =CHCH3 of (E)-isomer), 4.22 (0.17H, dd, J = 6.3, 2.0 Hz, -CH=CHH of (Z)-isomer), 4.29 (0.83H, dd, J = 6.3, 2.0 Hz, -CH=CHH of (E)-isomer), 4.57 (0.17H, dd, J = 13.7, 2.0 Hz, -CH=CHH of (Z)-isomer), 4.59 (0.83H, dd, J = 13.7, 2.0 Hz, -CH=CHH of (E)-isomer), 5.93 (0.17H, dq, J = 11.0, 6.5 Hz, -CH=CHCH3 of (Z)-isomer), 5.98 (0.17H, br d, J = 11.0 Hz, ArCH=CHCH3 of (Z)-isomer), 6.11 (0.83H, br dq, J = 16.0, 1.6 Hz, ArCH=CHCH3 of (E)-isomer), 6.34 (0.83H, dq, J = 15.8, 6.8 Hz, -CH=CHCH3 of (E)-isomer), 6.49 (0.17H, dd, J = 13.7, 6.3 Hz, -OCH=CH2 of (Z)-isomer), 6.55 (0.83H, dd, J = 13.7, 6.3 Hz, -OCH=CH2 of (E)-isomer), 7.07 (2H, br d, J = 7.0 Hz, Ph-H), 7.23–7.37 (3H, m, Ph-H), 7.33 (1H, br s, pyrazole-H); 13C NMR (150 MHz, CDCl3): δ 16.0 (minor), 19.3, 53.4 (minor), 54.0, 91.9 (minor), 92.3, 114.7 (minor), 115.9, 126.5, 126.9, 127.7, 128.7, 128.8, 129.0 (minor), 131.5, 134.2 (minor), 136.9, 138.5 (minor), 150.4 (minor), 150.5; HREIMS m/z calcd. for C15H16N2O (M+) 240.1263, found 240.1256.
(E/Z)-4-(2-Methoxy)ethoxy-3-(1-propenyl)-2H-2-tritylpyrazole (15): oil; IR (film) vmax 1492 (C=C), 1446 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 1.39 (3H, d, J = 6.7 Hz, =CHCH3), 3.65 (0.5H, br t, J = 4.1 Hz, -OCH2CH2Br), 3.7 (1.5H, br t, J = 4.1 Hz, -CH2CH2Br), 4.07 (0.5H, br t, J = 3.9 Hz, -CH2CH2Br), 3.70 (1.5H, br t, J = 4.1 Hz, -OCH2CH2Br), 5.44 (1H, br d, J = 15.8 Hz, (E)-ArCH=CH-), 6.09–6.18 (1H, m, -CH=CHCH3), 7.11–7.20 (6H, m, Tr-H), 7.24–7.29 (9H, m, Tr-H), 7.32 (1H, s, pyrazole-H); 13C NMR (150 MHz, CDCl3): δ 18.9, 59.2, 70.6, 71.3, 79.0, 119.9, 124.8, 127.3, 127.4, 127.6, 130.1, 130.4, 142.9, 143.7; HREIMS m/z calcd. for C28H28N2O2 (M+) 424.2151, found 424.2157.
4-(2-Methoxy)ethoxy-3-(2-propenyl)-2H-2-tritylpyrazole (16): oil; IR (film) vmax 1580 (C=C), 1447 (C=C) cm−1; 1H NMR (600 MHz, CDCl3): δ 2.84 (2H, br d, J = 6.5 Hz, ArCH2CH=), 3.41 (3H, s, -OCH3), 3.66 (1H, br t, J = 5.0 Hz, -OCH2CH2O-), 4.05 (2H, br t, J = 5.0 Hz, -OCH2CH2O-), 4.60 (1H, dq, J = 17.0, 1.7 Hz, -CH=CHH), 4.64 (1H, dq, J = 10.5, 1.5 Hz, -CH=CHH), 7.10–7.13 (6H, m, Tr-H), 7.23–7.33 (9H, m, Tr-H), 7.34 (1H, s, pyrazole-H); 13C NMR (150 MHz, CDCl3): δ 31.2, 59.2, 71.2, 71.4, 78.5, 115.6, 125.5, 127.3, 127.6, 127.9, 130.1, 132.6, 143.0, 144.4; HREIMS m/z calcd. for C28H28N2O2 (M+) 424.2151, found 424.2157.
4.8. RCM of 13a and 14a and 14b
The RCM reactions of
13a and
14a and
14b in
Scheme 2 were carried out as described above.
1,7-Dihydro-1-tritylpyrano[3,2-c]pyrazole (8a): oil; IR (film) vmax 1583 (C=C), 1493 (C=C), 1446 (C=C) cm−1; 1H NMR (600 MHz, CDCl3): δ 2.27 (2H, dd, J = 3.5, 2.0 Hz, ArCH2CH=CH-), 4.49 (1H, dt, J = 6.5, 3.5 Hz, -OCH=CHCH2-), 6.33 (1H, dt, J = 6.4, 2.0 Hz, -OCH=CHCH2-), 7.12–7.15 (6H, m, Tr-H), 7.26–7.32 (9H, m, Tr-H), 7.32 (1H, s, pyrazole-H); 13C NMR (150 MHz, CDCl3): δ 22.5, 29.7, 78.6, 98.2, 124.3, 127.6, 127.6, 127.9, 130.4, 140.3, 142.6; HREIMS m/z calcd. for C25H20N2O (M+) 364.1575, found 364.1576.
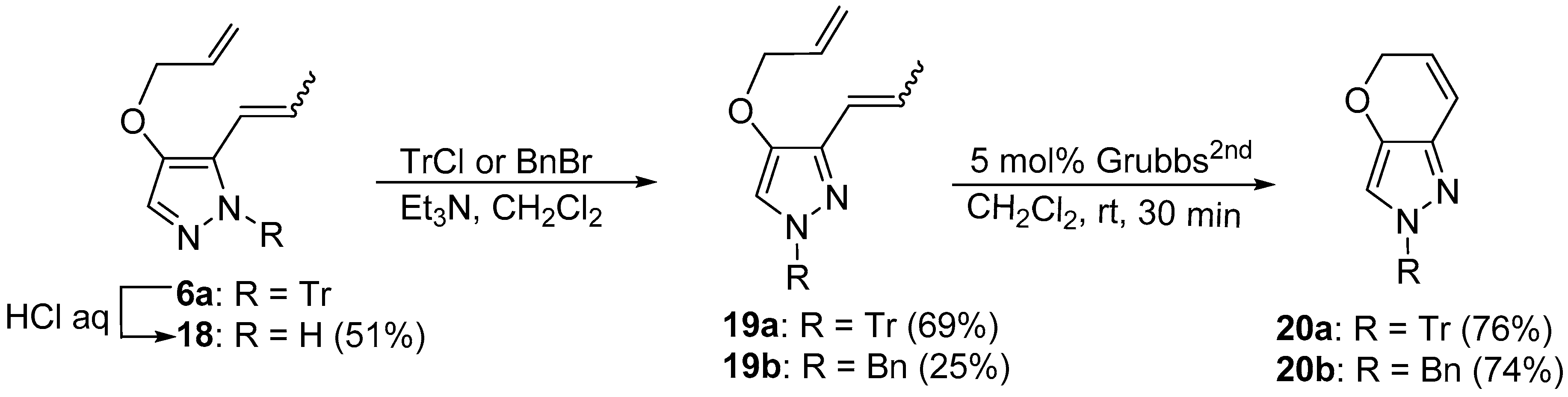
4.9. Acid-Catalyzed Hydrolysis of 6a (Scheme 3)
To a solution of 6a (121.5 mg, 0.30 mmol) in acetone (10 mL) was added 1 N aqueous HCl (0.6 mL). The reaction mixture was warmed under reflux for 90 min with stirring. After the reaction mixture had cooled, it was treated with saturated aqueous NaHCO3 and extracted with CH2Cl2. The separated organic layer was dried over MgSO4, filtered, and condensed under reduced pressure to give a crude residue, which was purified using silica gel column chromatography (eluent: EtOAc:hexane = 1:2) to afford (Z)-18 (9.1 mg, 20%) and (E)-18 (14.2 mg, 31%).
(E)-4-Allyloxy-5-(1-propenyl)-1H-pyrazole ((E)-18): oil; IR (film) vmax 1568 (C=C), 1516 (C=C) cm−1; 1H NMR (600 MHz, CDCl3): δ 1.89 (3H, br d, J = 6.0 Hz, CH3CH=), 4.60 (2H, dt, J = 5.5, 1.5 Hz, -OCH2CH=CH2), 4.46 (1H, dq, J = 10.0, 1.5 Hz, -CH=CHH), 5.40 (1H, dq, J = 17.3, 1.5 Hz, -CH=CHH), 6.04 (1H, ddt, J = 17.3, 10.5, 5.5 Hz, -OCH2CH=CH2), 6.34 (1H, d, J = 16.7 Hz, ArCH=CH-), 6.35–6.41 (1H, m, -CH=CHCH3), 7.22 (1H, s, pyrazole-H); 13C NMR (150 MHz, CDCl3): δ 18.9, 72.7, 117.7, 118.7, 127.6, 133.4, 142.0; HREIMS m/z calcd. for C9H12N2O (M+) 164.0950, found 164.0950.
(Z)-4-Allyloxy-5-(1-propenyl)-1H-pyrazole ((Z)-18): oil; IR (film) vmax 1570 (C=C), 1524 (C=C), 1450 (C=C) cm−1; 1H NMR (600 MHz, CDCl3): δ 1.98 (3H, dd, J = 7.0, 1.8 Hz, CH3CH=), 3.49 (3H, s, -OCH3), 4.45 (2H, dt, J = 5.2, 1.5 Hz, -OCH2CH=CH2), 5.27 (1H, dq, J = 10.6, 1.5 Hz, -CH=CHH), 5.38 (1H, dq, J = 17.0, 1.5 Hz, -CH=CHH), 5.82 (1H, dq, J = 11.4, 7.0 Hz, -CH=CHCH3), 6.03 (1H, ddt, J = 17.3, 10.6, 5.3 Hz, -OCH2CH=CH2), 6.27 (1H, dq, J = 11.5, 1.5 Hz, ArCH=CHCH3), 7.27 (1H, s, pyrazole-H); 13C NMR (150 MHz, CDCl3): δ 69.1, 69.3, 117.7, 118.7, 127.6, 133.4, 142.0; HREIMS m/z calcd. for C9H12N2O (M+) 164.0950, found 164.0949.
4.10. Reprotection of 18 (Scheme 3)
General procedure: To a stereo mixture of (E/Z)-18 (15.9 mg, 0.10 mmol) in CH2Cl2 (10 mL) were added TrCl (43.0 mg, 0.15 mmol) and Et3N (0.022 mL, 0.15 mmol) at 0 °C. The reaction mixture was stirred at rt overnight, and then quenched with saturated aqueous NH4Cl and extracted with CH2Cl2. The organic layer was dried over MgSO4, filtered, and condensed under reduced pressure to give a crude residue, which was purified using silica gel column chromatography (eluent: EtOAc:hexane = 1:4) to afford 19a (28.9 mg, 68%) as an oil.
(E/Z)-4-Allyloxy-3-(1-propenyl)-1H-1-tritylpyrazole (19a): oil; IR (film) vmax 1560 (C=C), 1491 (C=C), 1445 (C=C) cm−1; 1H NMR of (E)-isomer (600 MHz, CDCl3): δ 1.90 (3H, dd, J = 7.1, 1.8 Hz, CH3CH=CH-), 4.27 (2H, dt, J = 5.6, 1.5 Hz, -OCH2CH=CH2), 5.20 (1H, ddd, J = 10.6, 3.2, 1.5 Hz, -CH2CH=CHH), 5.28 (1H, ddd, J = 17.0, 3.2, 1.8 Hz, -CH2CH=CHH), 5.75 (1H, dq, J = 11.5, 7.1 Hz, -CH=CHCH3), 5.95 (1H, ddt, J = 17.3, 10.7, 5.6 Hz, -OCH2CH=CH2), 6.29 (1H, dq, J = 11.5, 1.5 Hz, ArCH=CHCH3), 6.84 (1H, s, pyrazole-H), 7.14–7.18 (6H, m, Tr-H), 7.26–7.30 (9H, m, Tr-H); 13C NMR (150 MHz, CDCl3): δ (14.2), 15.6, (60.4), 72.9, 78.6, (117.4), 117.6, 117.7, 127.4, 127.5, (127.6), 127.9, 130.4, 133.3, (138.6), (142.0), 143.4, signals in parentheses correspond to some of those of the (Z)-isomer; HREIMS m/z calcd. for C28H26N2O (M+) 406.2045, found 406.2040.
(E)-4-Allyloxy-1-benzyl-3-(1-propenyl)-1H-pyrazole (19b): oil; IR (film) vmax 1566 (C=C), 1496 (C=C), 1445 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 1.87 (3H, dd, J = 6.3, 1.2 Hz, CH3CH=CH-), 4.35 (2H, dt, J = 5.4, 1.5 Hz, -OCH2CH=CH2), 5.16 (2H, s, ArCH2Ph), 5.24 (1H, dq, J = 10.5, 1.5 Hz, -CH2CH=CHH), 5.36 (1H, dq, J = 17.2, 1.6 Hz, -CH2CH=CHH), 6.00 (1H, ddt, J = 17.2, 10.5, 5.4 Hz, -OCH2CH=CH2), 6.40 (1H, br d, J = 16.3 Hz, ArCH=CHCH3), 6.53 (1H, dq, J = 16.3, 6.3 Hz, ArCH=CHCH3), 6.92 (1H, s, pyrazole-H), 7.18 (2H, br d, J = 8.0 Hz, Ph-H), 7.26–7.35 (3H, m, Ph-H); 13C NMR (100 MHz, CDCl3): δ 18.9, 56.5, 72.6, 114.7, 117.6, 121.4, 127.4, 127.9, 128.7, 133.2, 136.8, 138.4, 143.1; HREIMS m/z calcd. for C16H18N2O (M+) 254.1419, found 254.1416.
4.11. RCM of 19
The RCM reactions of 19 were carried out in a similar manner to that described above to afford 20.
2,5-Dihydro-2-tritylpyrano[3,2-c]pyrazole (20a): oil; IR (film) vmax 1492 (C=C), 1447 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 4.76 (2H, dd, J = 2.9, 1.9 Hz, -OCH2CH=CH-), 5.72 (1H, dt, J = 10.2, 3.5 Hz, -OCH2CH=CH-), 6.62 (1H, br d, J = 10.0 Hz, -OCH2CH=CHAr), 6.80 (1H, s, pyrazole-H), 7.18–7.20 (6H, m, Tr-H), 7.28–7.31 (9H, m, Tr-H); 13C NMR (150 MHz, CDCl3): δ 67.2, 78.0, 116.5, 120.1, 122.7, 126.5, 127.6, 127.7, 137.5, 139.1, 143.3; HREIMS m/z calcd. for C25H20N2O (M+) 364.1575, found 364.1584.
2-Benzyl-2,5-dihydropyrano[3,2-c]pyrazole (20b): oil; IR (film) vmax 1660 (C=C), 1576 (C=C) cm−1; 1H NMR (400 MHz, CDCl3): δ 4.77 (2H, dd, J = 3.5, 1.9 Hz, -OCH2CH=CH-), 5.73 (1H, dt, J = 10.0, 3.5 Hz, -OCH2CH=CH-), 6.63 (1H, dt, J = 10.0, 1.9 Hz, -OCH2CH=CHAr), 6.84 (1H, s, pyrazole-H), 7.18–7.20 (2H, br d, J = 6.5 Hz, Ph-H), 7.27–7.36 (3H, m, Ph-H); 13C NMR (100 MHz, CDCl3): δ 56.4, 67.2, 113.3, 119.5, 122.3, 127.5, 128.0, 128.8, 136.6, 137.2, 140.5; HREIMS m/z calcd. for C13H12N2O (M+) 212.0949, found 212.0950.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
