
Synthesis and Telomeric G-Quadruplex-Stabilizing Ability of Macrocyclic Hexaoxazoles Bearing Three Side Chains

 ,
,
Abstract
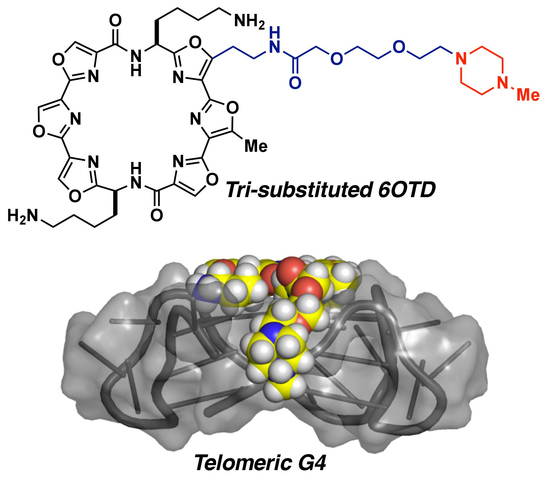
:
1. Introduction
2. Results and Discussion
2.1. Design of 6OTD Derivatives for Targeting Telomeric G4
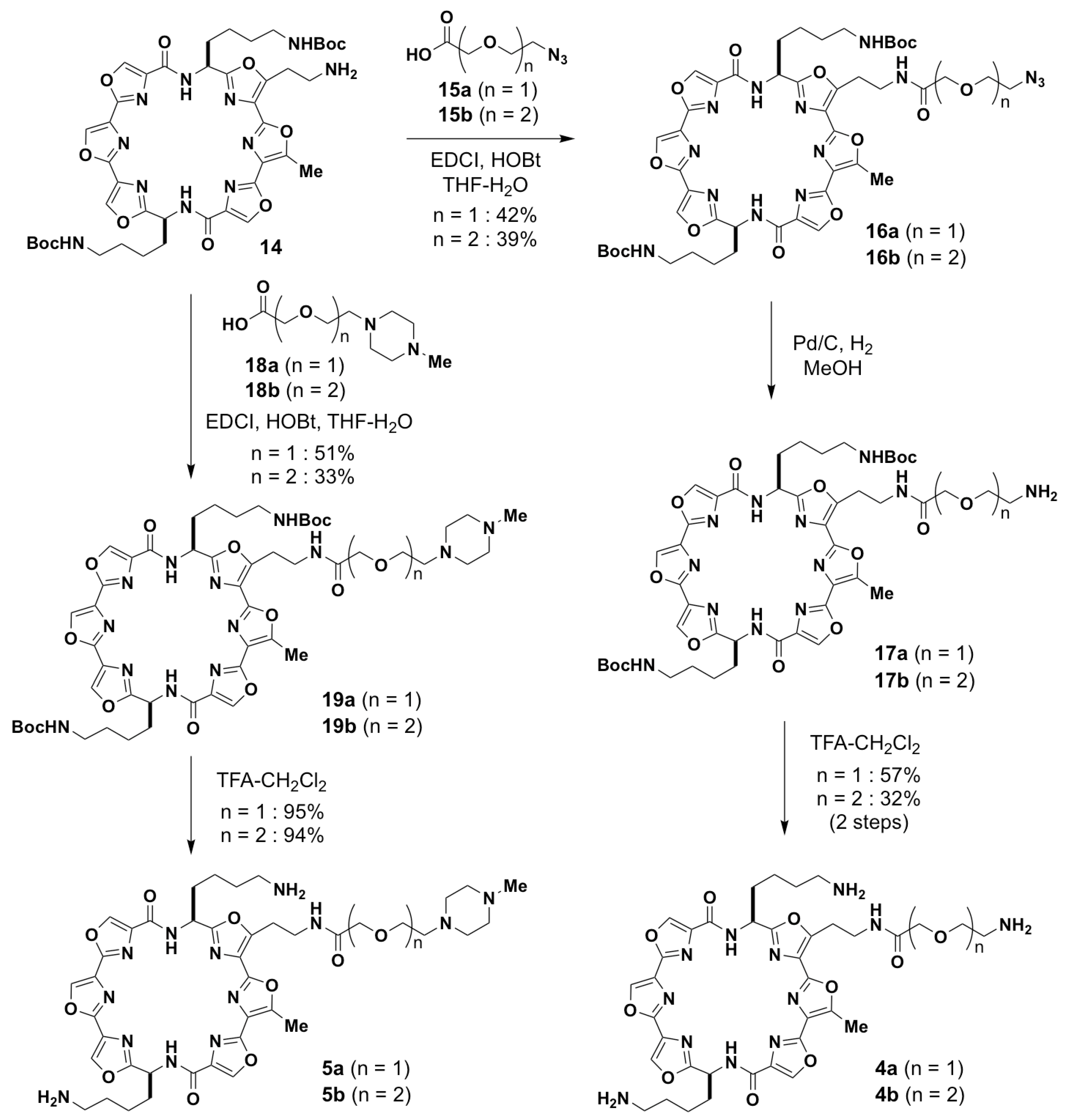
2.2. Synthesis of Tri-Substituted 6OTD Derivatives 3–5
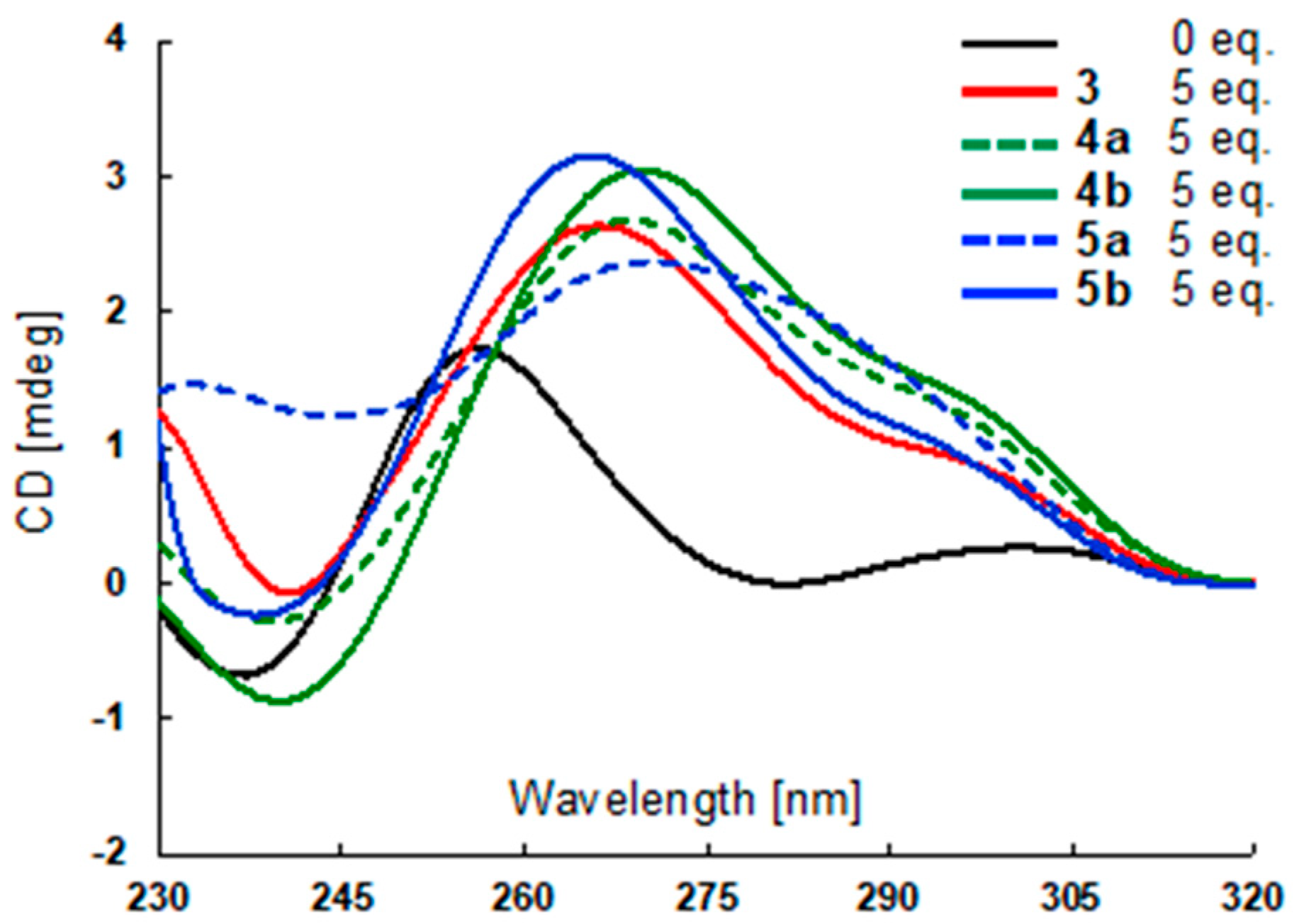
2.3. Stabilization Abilities of 3–5 for G4-Forming DNA Sequences by Fluorescence Resonance Energy Transfer (FRET) Melting Analysis
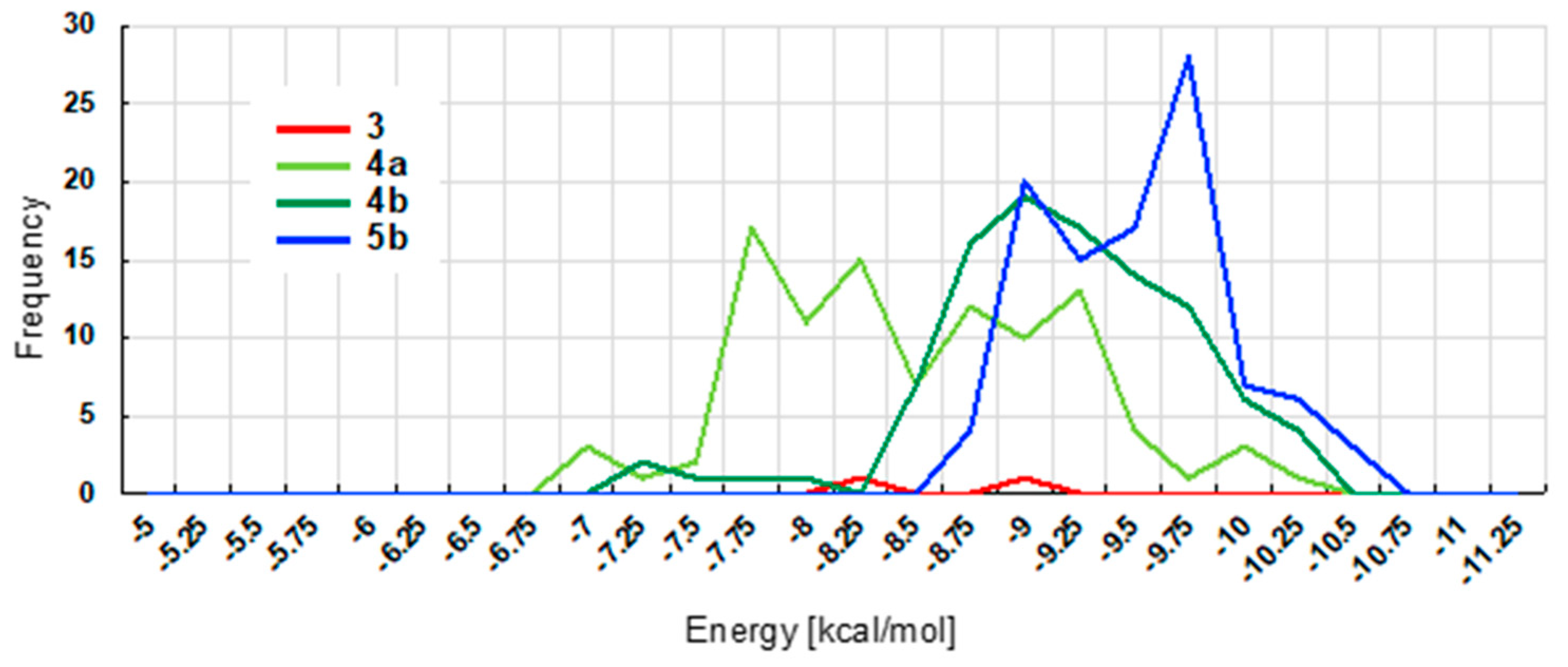
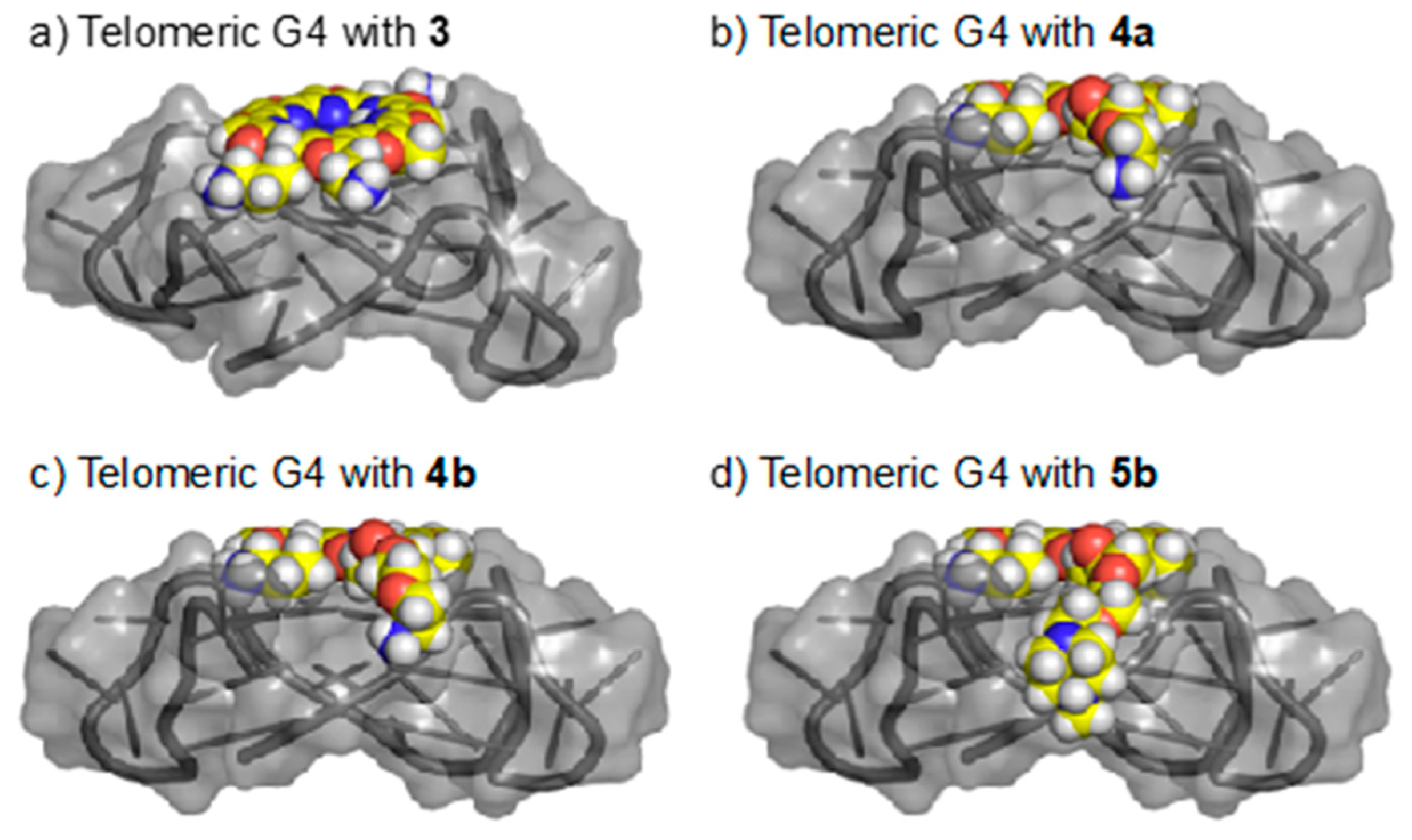
2.4. Docking Studies of Ligands 3, 4, and 5b with Telomeric G4
3. Materials and Methods
3.1. General
3.2. Synthesis Methods
3.3. FRET Melting Analysis
3.4. CD Spectrometry
3.5. Computational Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References and Notes
- Davis, J.T. G-quartets 40 years later: From 5’-GMP to molecular biology and supramolecular chemistry. Angew. Chem. Int. Ed. 2004, 43, 668–698. [Google Scholar] [CrossRef] [PubMed]
- Todd, A.K.; Johnston, M.; Neidle, S. Highly prevalent putative quadruplex sequence motifs in human DNA. Nucleic Acids Res. 2005, 33, 2901–2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huppert, J.L.; Balasubramanian, S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005, 33, 2908–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huppert, J.L.; Balasubramanian, S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007, 35, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.L.; Bugaut, A.; Kumari, S.; Balasubramanian, S. G-quadruplexes: The beginning and end of UTRs. Nucleic Acids Res. 2008, 36, 6260–6268. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Yadav, V.K.; Basundra, R.; Kumar, A.; Chowdhury, S. Evidence of genome-wide G4 DNA-mediated gene expression in human cancer cells. Nucleic Acids Res. 2009, 37, 4194–4204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, V.S.; Marsico, G.; Boutell, J.M.; Di Antonio, M.; Smith, G.P.; Balasubramanian, S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015, 33, 877–881. [Google Scholar] [CrossRef] [Green Version]
- Paeschke, K.; Capra, J.A.; Zakian, V.A. DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell 2011, 145, 678–691. [Google Scholar] [CrossRef]
- Kanoh, Y.; Matsumoto, S.; Fukatsu, R.; Kakusho, N.; Kono, N.; Renard-Guillet, C.; Masuda, K.; Iida, K.; Nagasawa, K.; Shirahige, K.; et al. Rif1 binds to G quadruplexes and suppresses replication over long distances. Nat. Struct. Mol. Biol. 2015, 22, 889–897. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.J.; Phan, A.T.; Kuryavyi, V. Human telomere, oncogenic promoter and 5’-UTR G-quadruplexes: Diverse higher order DNA and RNA targets for cancer therapeutics. Nucleic Acids Res. 2007, 35, 7429–7455. [Google Scholar] [CrossRef] [PubMed]
- Lipps, H.J.; Rhodes, D. G-quadruplex structures: In vivo evidence and function. Trends Cell Biol. 2009, 19, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Bugaut, A.; Balasubramanian, S. 5′-UTR RNA G-quadruplexes: Translation regulation and targeting. Nucleic Acids Res. 2012, 40, 4727–4741. [Google Scholar] [CrossRef] [PubMed]
- Le, D.D.; Di Antonio, M.; Chan, L.K.; Balasubramanian, S. G-quadruplex ligands exhibit differential G-tetrad selectivity. Chem. Commun. 2015, 51, 8048–8050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjan, N.; Davis, E.; Xue, L.; Arya, D.P. Dual recognition of the human telomeric G-quadruplex by a neomycin-anthraquinone conjugate. Chem. Commun. 2013, 49, 5796–5798. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.Y.; Jia, G.Q.; Zhou, J.; Han, G.Y.; Li, C. Evidence for the binding mode of porphyrins to G-quadruplex DNA. Phys. Chem. Chem. Phys. 2009, 11, 4025–4032. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.T.; Kuryavyi, V.; Gaw, H.Y.; Patel, D.J. Small-molecule interaction with a five-guanine-tract G-quadruplex structure from the human MYC promoter. Nat. Chem. Biol. 2005, 1, 167–173. [Google Scholar] [CrossRef] [Green Version]
- Chung, W.J.; Heddi, B.; Hamon, F.; Teulade-Fichou, M.P.; Phan, A.T. Solution structure of a G-quadruplex bound to the bisquinolinium compound Phen-DC3. Angew. Chem. Int. Ed. 2014, 53, 999–1002. [Google Scholar] [CrossRef]
- Marchand, A.; Granzhan, A.; Iida, K.; Tsushima, Y.; Ma, Y.; Nagasawa, K.; Teulade-Fichou, M.P.; Gabelica, V. Ligand-Induced Conformational Changes with Cation Ejection upon Binding to Human Telomeric DNA G-Quadruplexes. J. Am. Chem. Soc. 2015, 137, 750–756. [Google Scholar] [CrossRef]
- Hamon, F.; Largy, E.; Guedin-Beaurepaire, A.; Rouchon-Dagois, M.; Sidibe, A.; Monchaud, D.; Mergny, J.L.; Riou, J.F.; Nguyen, C.H.; Teulade-Fichou, M.P. An Acyclic Oligoheteroaryle That Discriminates Strongly between Diverse G-Quadruplex Topologies. Angew. Chem. Int. Ed. 2011, 50, 8745–8749. [Google Scholar] [CrossRef]
- Martino, L.; Virno, A.; Pagano, B.; Virgilio, A.; Di Micco, S.; Galeone, A.; Giancola, C.; Bifulco, G.; Mayol, L.; Randazzo, A. Structural and thermodynamic studies of the interaction of distamycin A with the parallel quadruplex structure [d(TGGGGT)]4. J. Am. Chem. Soc. 2007, 129, 16048–16056. [Google Scholar] [CrossRef]
- Iida, K.; Nagasawa, K. Macrocyclic Polyoxazoles as G-Quadruplex Ligands. Chem. Rec. 2013, 13, 539–548. [Google Scholar] [CrossRef]
- Kim, M.Y.; Vankayalapati, H.; Shin-Ya, K.; Wierzba, K.; Hurley, L.H. Telomestatin, a potent telomerase inhibitor that interacts quite specifically with the human telomeric intramolecular g-quadruplex. J. Am. Chem. Soc. 2002, 124, 2098–2099. [Google Scholar] [CrossRef] [PubMed]
- Shin-ya, K.; Wierzba, K.; Matsuo, K.; Ohtani, T.; Yamada, Y.; Furihata, K.; Hayakawa, Y.; Seto, H. Telomestatin, a novel telomerase inhibitor from Streptomyces anulatus. J. Am. Chem. Soc. 2001, 123, 1262–1263. [Google Scholar] [CrossRef]
- Sakuma, M.; Ma, Y.; Tsushima, Y.; Iida, K.; Hirokawa, T.; Nagasawa, K. Design and synthesis of unsymmetric macrocyclic hexaoxazole compounds with an ability to induce distinct G-quadruplex topologies in telomeric DNA. Org. Biomol. Chem. 2016, 14, 5109–5116. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Tsushima, Y.; Sakuma, M.; Sasaki, S.; Iida, K.; Okabe, S.; Seimiya, H.; Hirokawa, T.; Nagasawa, K. Development of G-quadruplex ligands for selective induction of a parallel-type topology. Org. Biomol. Chem. 2018, 16, 7375–7382. [Google Scholar] [CrossRef]
- Teulade-Fichou, M.P.; Carrasco, C.; Guittat, L.; Bailly, C.; Alberti, P.; Mergny, J.L.; David, A.; Lehn, J.M.; Wilson, W.D. Selective recognition of G-qQuadruplex telomeric DNA by a bis(quinacridine) macrocycle. J. Am. Chem. Soc. 2003, 125, 4732–4740. [Google Scholar] [CrossRef] [PubMed]
- Di Leva, F.S.; Zizza, P.; Cingolani, C.; D’Angelo, C.; Pagano, B.; Amato, J.; Salvati, E.; Sissi, C.; Pinato, O.; Marinelli, L.; et al. Exploring the chemical space of G-quadruplex binders: Discovery of a novel chemotype targeting the human telomeric sequence. J. Med. Chem. 2013, 56, 9646–9654. [Google Scholar] [CrossRef]
- Maji, B.; Kumar, K.; Kaulage, M.; Muniyappa, K.; Bhattacharya, S. Design and synthesis of new benzimidazole-carbazole conjugates for the stabilization of human telomeric DNA, telomerase inhibition, and their selective action on cancer cells. J. Med. Chem. 2014, 57, 6973–6988. [Google Scholar] [CrossRef]
- Lavrado, J.; Ohnmacht, S.A.; Correia, I.; Leitao, C.; Pisco, S.; Gunaratnam, M.; Moreira, R.; Neidle, S.; Santos, D.J.; Paulo, A. Indolo [3,2-c]quinoline G-quadruplex stabilizers: A structural analysis of binding to the human telomeric G-quadruplex. Chem. Med. Chem. 2015, 10, 836–849. [Google Scholar] [CrossRef]
- Abraham Punnoose, J.; Ma, Y.; Li, Y.; Sakuma, M.; Mandal, S.; Nagasawa, K.; Mao, H. Adaptive and Specific Recognition of Telomeric G-Quadruplexes via Polyvalency Induced Unstacking of Binding Units. J. Am. Chem. Soc. 2017, 139, 7476–7484. [Google Scholar] [CrossRef] [PubMed]
- Pennarun, G.; Granotier, C.; Gauthier, L.R.; Gomez, D.; Boussin, F.D. Apoptosis related to telomere instability and cell cycle alterations in human glioma cells treated by new highly selective G-quadruplex ligands. Oncogene 2005, 24, 2917–2928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruden, M.; Puri, N. Novel anticancer therapeutics targeting telomerase. Cancer Treat. Rev. 2013, 39, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y. Chemistry in human telomere biology: Structure, function and targeting of telomere DNA/RNA. Chem. Soc. Rev. 2011, 40, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Neidle, S. G-quadruplex nucleic acids as therapeutic targets. Curr. Opin. Chem. Biol. 2009, 13, 345–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansel-Hertsch, R.; Di Antonio, M.; Balasubramanian, S. DNA G-quadruplexes in the human genome: Detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.B.; Hu, M.H.; Liu, G.C.; Wang, J.; Ou, T.M.; Gu, L.Q.; Huang, Z.S.; Tan, J.H. Visualization of NRAS RNA G-Quadruplex Structures in Cells with an Engineered Fluorogenic Hybridization Probe. J. Am. Chem. Soc. 2016, 138, 10382–10385. [Google Scholar] [CrossRef]
- Hu, M.H.; Chen, S.B.; Wang, B.; Ou, T.M.; Gu, L.Q.; Tan, J.H.; Huang, Z.S. Specific targeting of telomeric multimeric G-quadruplexes by a new triaryl-substituted imidazole. Nucleic Acids Res. 2017, 45, 1606–1618. [Google Scholar] [CrossRef]
- Hu, M.H.; Wang, Y.Q.; Yu, Z.Y.; Hu, L.N.; Ou, T.M.; Chen, S.B.; Huang, Z.S.; Tan, J.H. Discovery of a New Four-Leaf Clover-Like Ligand as a Potent c-MYC Transcription Inhibitor Specifically Targeting the Promoter G-Quadruplex. J. Med. Chem. 2018, 61, 2447–2459. [Google Scholar] [CrossRef]
- Chung, W.J.; Heddi, B.; Tera, M.; Iida, K.; Nagasawa, K.; Phan, A.T. Solution structure of an intramolecular (3 + 1) human telomeric G-quadruplex bound to a telomestatin derivative. J. Am. Chem. Soc. 2013, 135, 13495–13501. [Google Scholar] [CrossRef]
- Iida, K.; Majima, S.; Ohtake, T.; Tera, M.; Shin-ya, K.; Nagasawa, K. Design and synthesis of g-quadruplex ligands bearing macrocyclic hexaoxazoles with four-way side chains. Heterocycles 2012, 84, 401–411. [Google Scholar]
- Tera, M.; Ishizuka, H.; Takagi, M.; Suganuma, M.; Shin-ya, K.; Nagasawa, K. Macrocyclic hexaoxazoles as sequence- and mode-selective G-quadruplex binders. Angew. Chem. Int. Ed. 2008, 47, 5557–5560. [Google Scholar] [CrossRef] [PubMed]
- De Cian, A.; Guittat, L.; Kaiser, M.; Sacca, B.; Amrane, S.; Bourdoncle, A.; Alberti, P.; Teulade-Fichou, M.P.; Lacroix, L.; Mergny, J.L. Fluorescence-based melting assays for studying quadruplex ligands. Methods 2007, 42, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Ou, T.M.; Lu, Y.J.; Tan, J.H.; Huang, Z.S.; Wong, K.Y.; Gu, L.Q. G-quadruplexes: Targets in anticancer drug design. ChemMedChem 2008, 3, 690–713. [Google Scholar] [CrossRef] [PubMed]
- Vorlickova, M.; Kejnovska, I.; Sagi, J.; Renciuk, D.; Bednarova, K.; Motlova, J.; Kypr, J. Circular dichroism and guanine quadruplexes. Methods 2012, 57, 64–75. [Google Scholar] [CrossRef] [PubMed]
- The sterically hindered side chains introduced in the compounds 4 and 5 might cause the topology change from hybrid into parallel structure by interfering with the loops.
- To evaluate the docking accuracy of our protocol, we carried out the re-docking study using the known x-ray structure of G-quadruplex-ligand complex (PDB ID: 3UYH). In the re-docking experiments, we obtained near native poses (RMSD < 3 Å) in top 100 candidates, however, correlation between the docking scores and the RMSD plots were low. To avoid the choices of the false positive poses from the best-rank only, we decided to consider 100 poses together with the docking scores. See Figure S2.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
Sample Availability: Samples of all compounds are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA | ΔTm Values (°C) | |||||
|---|---|---|---|---|---|---|
| L2H2-6M(2)OTD (1) | 3 | 4a | 4b | 5a | 5b | |
| telo21 | 18.1 ± 0.0 | 23.4 ± 0.1 | 23.5 ± 0.2 | 24.4 ± 0.5 | 18.0 ± 0.9 | 26.2 ± 0.3 |
| c-kit | 18.3 ± 0.4 | 20.8 ± 0.2 | 19.5 ± 0.3 | 20.7 ± 0.1 | 12.9 ± 0.6 | 12.1 ± 0.3 |
| K-ras | 10.3 ± 0.4 | 13.0 ± 1.0 | 11.9 ± 0.2 | 13.5 ± 0.2 | 9.0 ± 0.2 | 8.2 ± 0.4 |
| dsDNA | 0.1 ± 0.0 | 0.0 ± 0.1 | 0.1 ± 0.0 | 0.0 ± 0.2 | 0.0 ± 0.1 | 0.0 ± 0.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Iida, K.; Sasaki, S.; Hirokawa, T.; Heddi, B.; Phan, A.T.; Nagasawa, K. Synthesis and Telomeric G-Quadruplex-Stabilizing Ability of Macrocyclic Hexaoxazoles Bearing Three Side Chains. Molecules 2019, 24, 263. https://doi.org/10.3390/molecules24020263
Ma Y, Iida K, Sasaki S, Hirokawa T, Heddi B, Phan AT, Nagasawa K. Synthesis and Telomeric G-Quadruplex-Stabilizing Ability of Macrocyclic Hexaoxazoles Bearing Three Side Chains. Molecules. 2019; 24(2):263. https://doi.org/10.3390/molecules24020263
Chicago/Turabian StyleMa, Yue, Keisuke Iida, Shogo Sasaki, Takatsugu Hirokawa, Brahim Heddi, Anh Tuân Phan, and Kazuo Nagasawa. 2019. "Synthesis and Telomeric G-Quadruplex-Stabilizing Ability of Macrocyclic Hexaoxazoles Bearing Three Side Chains" Molecules 24, no. 2: 263. https://doi.org/10.3390/molecules24020263