
Studies of Halogen Bonding Induced by Pentafluorosulfanyl Aryl Iodides: A Potential Group of Halogen Bond Donors in a Rational Drug Design


Abstract
:
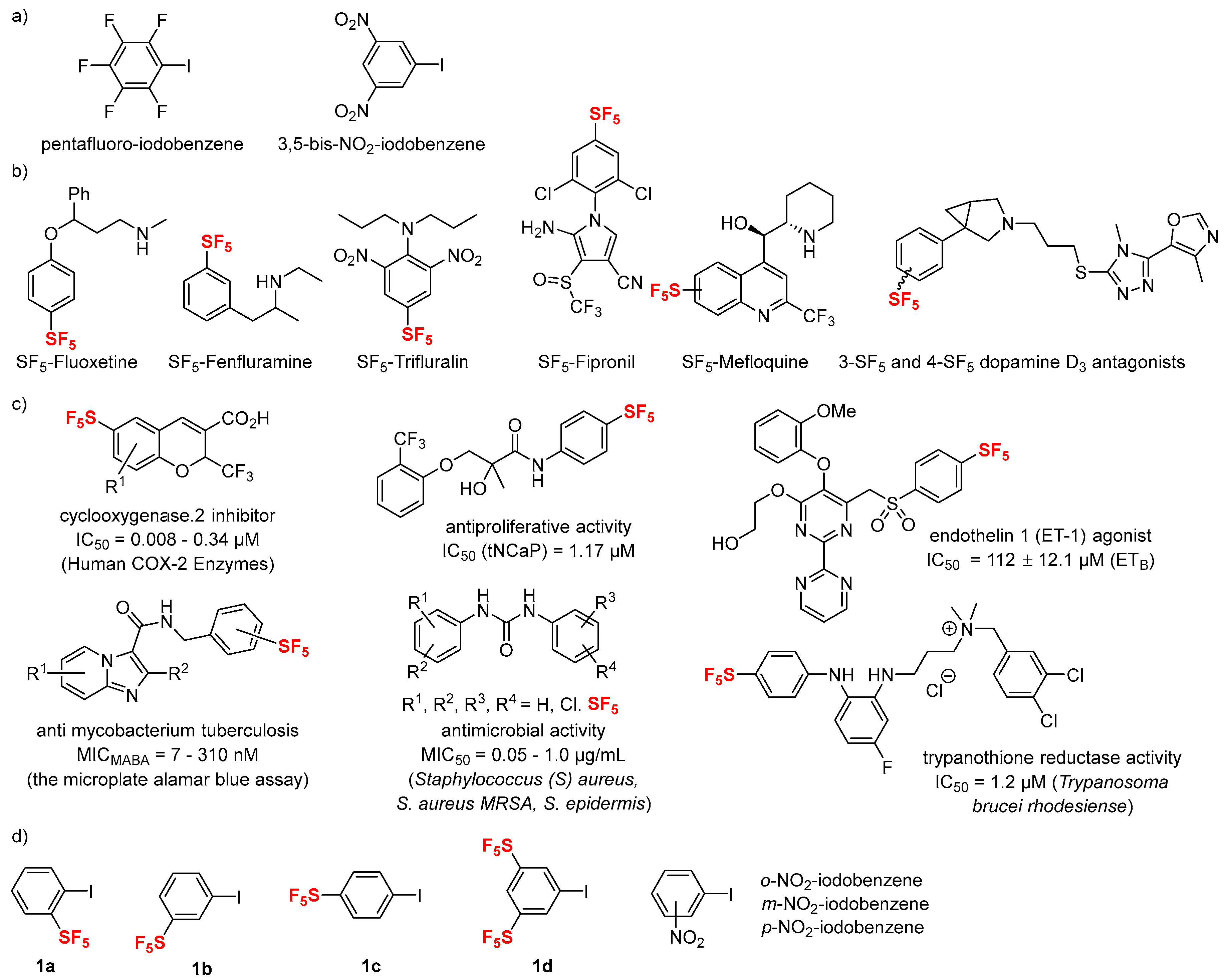
1. Introduction
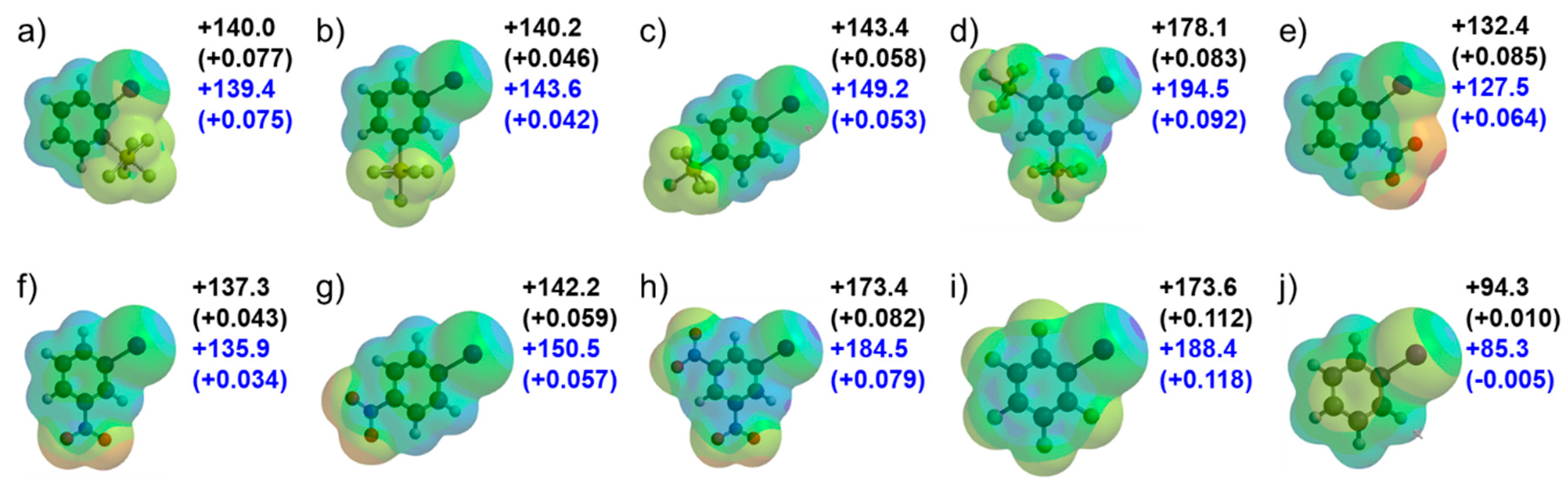
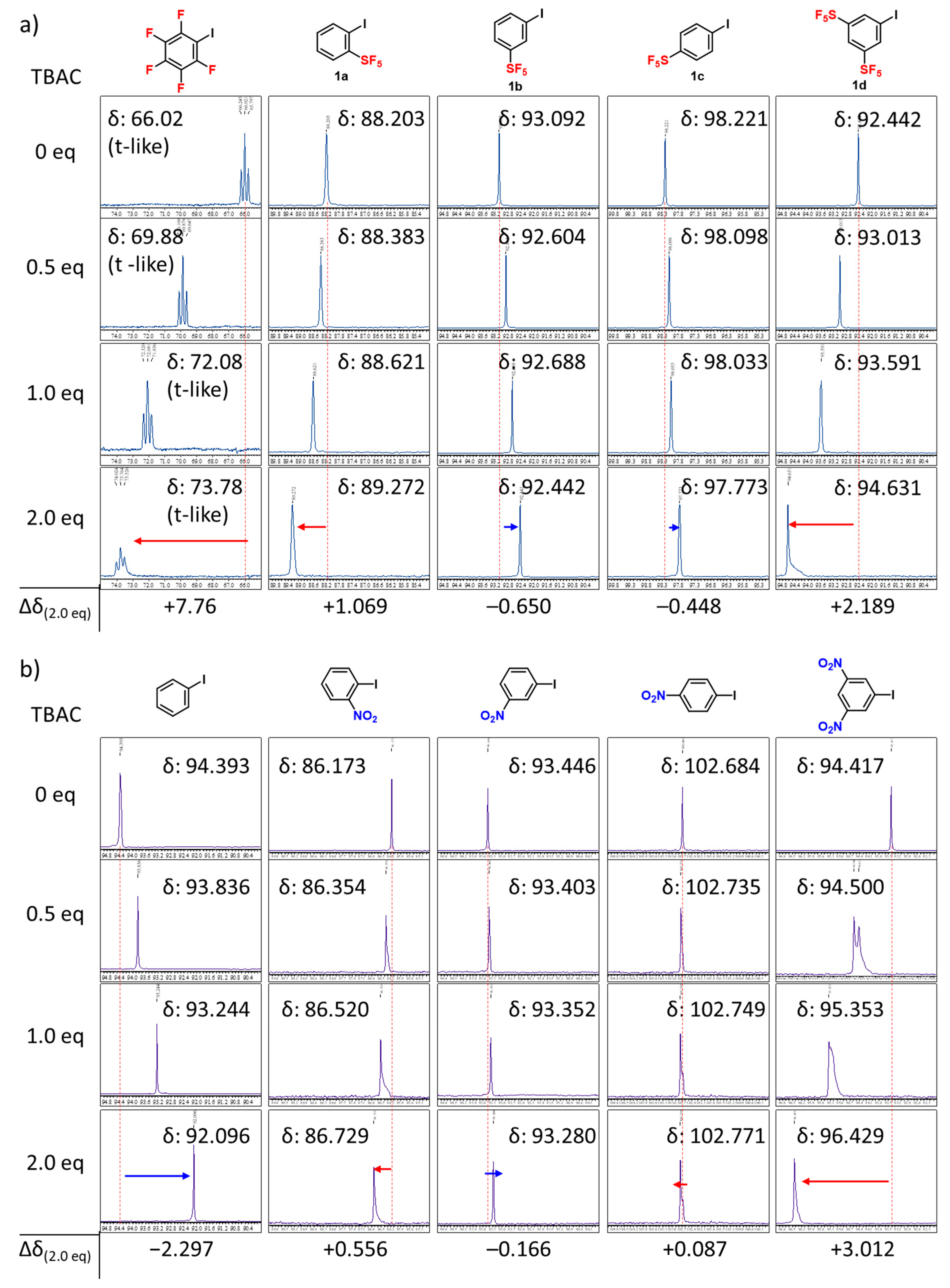
2. Results and Discussion
3. Materials and Methods
3.1. General Procedure of Pentafluoro-λ6-Sulfanyl Iodobenzene

3.1.1. Pentafluoro(2-iodophenyl)-λ6-sulfane (1a)
3.1.2. Pentafluoro(3-iodophenyl)-λ6-sulfane (1b)
3.1.3. Pentafluoro(4-iodophenyl)-λ6-sulfane (1c)
3.1.4. (5-Iodo-1,3-phenylene)bis(pentafluoro-λ6-sulfane) (1d)
3.2. Computational Method
3.2.1. Calculations for Electrostatic Potential Values
3.2.2. Calculations for Interaction Energies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulfield, D.; Huber, S.M. Halogen Bonding in Organic Synthesis and Organocatalysis. Chem. Eur. J. 2016, 22, 14434–14450. [Google Scholar] [CrossRef] [PubMed]
- Tepper, R.; Schubert, U.S. Halogen Bonding in Solution: Anion Recognition, Templated Self-Assembly, and Organocatalysis. Angew. Chem. Int. Ed. 2018, 57, 6004–6016. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bisoyi, H.K.H.; Urbas, A.M.; Bunning, T.J.; Li, Q. Halogen Bond: An Emerging Supramolecular Tool in the Design of Functional Mesomorphic Materials. Chem. Eur. J. 2019, 25, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Farina, A.; Meille, S.V.; Messina, M.T.; Metrangolo, P.; Resnati, G.; Vecchio, G. Resolution of Racemic 1,2-Dibromohexafluoropropane through Halogen-Bonded Supramolecular Helices. Angew. Chem. Int. Ed. 1999, 38, 2433–2436. [Google Scholar] [CrossRef]
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.; Murray, J.S. An Overview of Halogen Bonding. J. Mol. Model 2007, 13, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murraya, J.S.; Clark, T. Halogen Bonding: An Electrostatically-Driven Highly Directional Noncovalent Interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Sarwar, M.G.; Dragisic, B.; Salsberg, L.J.; Gouliaras, C.; Taylor, M.S. Thermodynamics of Halogen Bonding in Solution: Substituent, Structural, and Solvent Effects. J. Am. Chem. Soc. 2010, 132, 1646–1653. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Wakisaka, A.; Ono, T.; Sonoda, T. Magnitude and Origin of the Attraction and Directionality of the Halogen Bonds of the Complexes of C6F5X and C6H5X (X = I, Br, Cl and F) with Pyridine. Chem. Eur. J. 2012, 18, 951–960. [Google Scholar] [CrossRef]
- Raatikainen, K.; Rissanen, K. Hierarchical halogen bonding induces polymorphism. Cryst. Eng. Comm. 2009, 11, 750–752. [Google Scholar] [CrossRef]
- Rosokha, S.V.; Loboda, E.A. Interplay of Halogen and π–π Charge-Transfer Bondings in Intermolecular Associates of Bromo- or Iododinitrobenzene with Tetramethyl-p-phenylenediamine. J. Phys. Chem. A 2015, 119, 3833–3842. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.Q.; Zhao, X.R.; Wang, H.; Jin, W.J. The Competition of σ-Hole···Cl− and π-Hole···Cl− Bonds between C6F5X (X = F, Cl, Br, I) and the Chloride Anion and Its Potential Application in Separation Science. J. Phys. Chem. B 2014, 118, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Viger-Gravel, J.; Leclerc, S.; Korobkov, I.; Bryce, D. Direct Investigation of Halogen Bonds by Solid-State Multinuclear Magnetic Resonance Spectroscopy and Molecular Orbital Analysis. J. Am. Chem. Soc. 2014, 136, 6929–6942. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.C.; Dougherty, D.A. The Cation-π Interaction. Chem. Rev. 1997, 97, 1303–1324. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.-Y.; Kojima, T.; Koide, T.; Tachikawa, M.; Hiraoka, S. A Balance between van der Waals and Cation–π Interactions Stabilizes Hydrophobic Assemblies. Chem. Eur. J. 2018, 24, 9130–9135. [Google Scholar] [CrossRef] [PubMed]
- Kozelka, J. Lone Pair–π Interactions in Biological Systems: Occurrence, Function, and Physical Origin. Eur. Biophys. J. 2017, 46, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Mooibroek, T.J.; Gamez, P.; Reedijk, J. Lone pair-π Interactions: A New Supramolecular bond? Cryst. Eng. Comm. 2008, 10, 1501–1515. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. The Bright Future of Unconventional σ/π-Hole Interactions. ChemPhysChem 2015, 16, 2496–2517. [Google Scholar] [CrossRef]
- Savastano, M.; García-Gallarín, C.; de la Torre, M.D.L.; Bazzicalupi, C.; Bianchi, A.; Melguizo, M. Anion-π and Lone Pair-π Interactions with s-Tetrazine-Based Ligands. Coord. Chem. Rev. 2019, 397, 112–137. [Google Scholar] [CrossRef]
- Mendez, L.; Henriquez, G.; Sirimulla, S.; Narayan, M. Looking Back, Looking Forward at Halogen Bonding in Drug Discovery. Molecules 2017, 22, 1397. [Google Scholar] [CrossRef]
- Parisini, E.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen Bonding in Halocarbon–Protein Complexes: A Structural Survey. Chem. Soc. Rev. 2011, 40, 2267–2278. [Google Scholar] [CrossRef] [PubMed]
- Scholfield, M.R.; Vander Zanden, V.M.; Carter, M.; Ho, P.S. Halogen Bonding (X-Bonding): A Biological Perspective. Protein Sci. 2013, 22, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Kolář, M.H.; Tabarrini, O. Halogen Bonding in Nucleic Acid Complexes. J. Med. Chem. 2017, 60, 8681–8690. [Google Scholar] [CrossRef]
- Lentz, D.; Seppelt, K.; Akiba, K.-Y. Chemistry of Hypervalent Compounds; Akiba, K.-Y., Ed.; Wiley-VCH: New York, NY, USA, 1999. [Google Scholar]
- Winter, R.W.; Dodean, R.A.; Gard, G.L. Fluorine Containing Synthons; Soloshonok, V.A., Ed.; American Chemical Society: Washington, DC, USA, 2005. [Google Scholar]
- Altomonte, S.; Zanda, M. Synthetic Chemistry and Biological Activity of Pentafluorosulphanyl (SF5) Organic Molecules. J. Fluorine Chem. 2012, 143, 57–93. [Google Scholar] [CrossRef]
- Kirsch, P.; Röschenthaler, G.-V. Current Fluoroorganic Chemistry, Vol. 949; American Chemical Society: Washington, DC, USA, 2007. [Google Scholar]
- Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH Verlag GmbH Co. KGaA: Weinheim, Germany, 2013. [Google Scholar]
- Savoie, P.R.; Welch, J.T. Preparation and Utility of Organic Pentafluorosulfanyl-Containing Compounds. Chem. Rev. 2015, 115, 1130–1190. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, P. The Petafluorosulfuranyl Group and Related Structures. In Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2004; pp. 146–156. [Google Scholar]
- Sheppard, W.A. Arylsulfur Pentafluorides. J. Am. Chem. Soc. 1962, 84, 3064–3072. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Unger, S.H.; Kim, K.H.; Nikaitani, D.; Lien, E.J. “Aromatic” Substituent Constants for Structure-Activity Correlations. J. Med. Chem. 1973, 16, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Muir, R.M.; Fujita, T.; Maloney, P.P.; Geiger, F.; Streich, M. The Correlation of Biological Activity of Plant Growth Regulators and Chloromycetin Derivatives with Hammett Constants and Partition Coefficients. J. Am. Chem. Soc. 1963, 85, 2817–2824. [Google Scholar] [CrossRef]
- Bégué, J.-P.; Bonnet-Delpon, D. Bioorganic and Medicinal Chemistry of Fluorine; Wiley: Hoboken, Germany, 2008. [Google Scholar]
- Westphal, M.V.; Wolfstädter, B.T.; Plancher, J.-M.; Gatfield, J.; Carreira, E.M. Evaluation of tert-Butyl Isosteres: Case Studies of Physicochemical and Pharmacokinetic Properties, Efficacies, and Activities. Chem. Med. Chem. 2015, 10, 461–469. [Google Scholar] [CrossRef]
- Savoie, P.R.; von Hahmann, C.N.; Penger, A.; Wei, Z.; Welch, J.T. The Control of Stereochemistry by the Pentafluorosulfanyl Group. Org. Biomol. Chem. 2018, 16, 3151–3159. [Google Scholar] [CrossRef]
- Joliton, A.; Plancher, J.-M.; Carreira, E.M. Formation of α-SF5-Enolate Enables Preparation of 3-SF5-Quinolin-2-ones, 3-SF5-Quinolines, and 3-SF5-Pyridin-2-ones: Evaluation of their Physicochemical Properties. Angew. Chem. Int. Ed. 2016, 55, 2113–2117. [Google Scholar] [CrossRef] [PubMed]
- Kokkonda, S.; Deng, X.; White, K.L.; Coteron, J.M.; Marco, M.; de las Heras, L.; White, J.; Mazouni, F.E.; Tomchick, D.R.; Manjalanagara, K.; et al. Tetrahydro-2-naphthyl and 2-Indanyl Triazolopyrimidines Targeting Plasmodium falciparum Dihydroorotate Dehydrogenase Display Potent and Selective Antimalarial Activity. J. Med. Chem. 2016, 59, 5416–5431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; He, C.; Liu, X.; Lu, Y.; Chen, T.; Pan, Q.; Xiong, J.; She, M.; Tu, Z.; et al. Pentafluorosulfanyl-Substituted Benzopyran Analogues as New Cyclooxygenase-2 Inhibitors with Excellent Pharmacokinetics and Efficacy in Blocking Inflammation. J. Med. Chem. 2017, 60, 4135–4146. [Google Scholar] [CrossRef] [PubMed]
- Moraski, G.C.; Bristol, R.; Seeger, N.; Boshoff, H.L.; Tsang, P.S.-Y.; Miller, M.J. Preparation and Evaluation of Potent Pentafluorosulfanyl Substituted Anti-Tuberculosis Compounds. Chem. Med. Chem. 2017, 12, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Sansook, S.; Ocasio, C.A.; Day, I.J.; Tizzard, G.J.; Coles, S.J.; Fedorov, O.; Bennett, J.M.; Elkins, J.M.; Spencer, J. Synthesis of kinase inhibitors containing a pentafluorosulfanyl moiety. Org. Biomol. Chem. 2017, 15, 8655–8660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sowaileh, M.F.; Hazlitt, R.A.; Colby, D.A. Application of the Pentafluorosulfanyl Group as a Bioisosteric Replacement. Chem. Med. Chem. 2017, 12, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Pujol, E.; Blanco-Cabra, N.; Julián, E.; Leiva, R.; Torrents, E.; Vázquez, S. Pentafluorosulfanyl-Containing Triclocarban Analogs with Potent Antimicrobial Activity. Molecules 2018, 23, 2853. [Google Scholar] [CrossRef] [PubMed]
- Pertusati, F.; Ferla, S.; Bassetto, M.; Brancale, A.; Khandil, S.; Westwell, A.D.; McGuigan, C. A New Series of Bicalutamide, Enzalutamide and Enobosarm Derivatives Carrying Pentafluorosulfanyl (SF5) and Pentafluoroethyl (C2F5) Substituents: Improved Antiproliferative Agents against Prostate Cancer. Eur. J. Med. Chem. 2019, 180, 1–14. [Google Scholar] [CrossRef]
- Yang, Y.-D.; Lu, X.; Tokunaga, E.; Shibata, N. 3,5-Bis(pentafluorosulfanyl)phenylboronic Acid: A New Organocatalyst for Conia-Ene Carbocyclization of 1,3-Dicarbonyl Compounds Having Terminal Alkynes. J. Fluor. Chem. 2012, 143, 204–209. [Google Scholar] [CrossRef]
- Yang, Y.-D.; Tokunaga, E.; Akiyama, H.; Saito, N.; Shibata, N. Bis(pentafluorosulfanyl)phenyl Azide as an Expeditious Tool for Click Chemistry toward Antitumor Pharmaceuticals. Chem. Med. Chem. 2014, 9, 913–917. [Google Scholar] [CrossRef]
- Iida, N.; Tokunaga, E.; Saito, N.; Shibata, N. Synthesis and Property of Novel Phthalocyanine Having a 3,5-Bis-pentafluorosulfanylphenyl Group on the α-Peripheral Position. J. Fluor. Chem. 2014, 168, 93–98. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Okuyama, K.; Tokunaga, E.; Saito, N.; Shiro, M.; Shibata, N. Synthesis of Diaryliodonium Salts Having Pentafluorosulfanylarenes and Their Application to Electrophilic Pentafluorosulfanylarylation of C-, O-, N-, and S-Nucleophiles. Org. Lett. 2015, 17, 3038–3041. [Google Scholar] [CrossRef] [PubMed]
- Iida, N.; Tanaka, K.; Tokunaga, E.; Mori, S.; Saito, N.; Shibata, N. Synthesis of Phthalocyanines with a Pentafluorosulfanyl Substituent at Peripheral Positions. Chem. Open 2015, 4, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Kosobokov, M.; Cui, B.; Balia, A.; Matsuzaki, K.; Tokunaga, E.; Saito, N.; Shibata, N. Importance of a Fluorine Substituent for the Preparation of meta- and para-Pentafluoro-λ6-sulfanyl-Substituted Pyridines. Angew. Chem. Int. Ed. 2016, 55, 10781–10785. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Takada, M.; Matsuzaki, K.; Saito, N.; Shibata, N. SF5-Pyridylaryl-λ3-iodonium Salts and Their Utility as Electrophilic Reagents to Access SF5-pyridine Derivatives in the Late-Stage of Synthesis. Chem. Commun. 2017, 53, 3850–3853. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Jia, S.; Tokunaga, E.; Saito, N.; Shibata, N. Silver-Induced Self-Immolative Cl–F Exchange Fluorination of Arylsulfur Chlorotetrafluorides: Synthesis of Arylsulfur Pentafluorides. Chem. Commun. 2017, 53, 12738–12741. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Tokunaga, E.; Shibata, N. Recent Advancements in the Synthesis of Pentafluorosulfanyl (SF5)-Containing Heteroaromatic Compounds. Tetrahedron Lett. 2017, 58, 4803–4815. [Google Scholar] [CrossRef]
- Das, P.; Takada, M.; Tokunaga, E.; Saito, N.; Shibata, N. Synthesis of Pyridine trans-Tetrafluoro-λ6-sulfane Derivatives via Radical Additions. Org. Chem. Front. 2018, 5, 719–724. [Google Scholar] [CrossRef]
- Das, P.; Niina, K.; Hiromura, T.; Tokunaga, E.; Saito, N.; Shibata, N. An Eccentric Rod-Like Linear Connection of Two Heterocycles: Synthesis of Pyridine trans-Tetrafluoro-λ6-sulfanyl Triazoles. Chem. Sci. 2018, 9, 4931–4936. [Google Scholar] [CrossRef]
- Cui, B.; Shibata, N. The Story of SF5-Substituted Pyridines. Phosphorus, Sulfur Silicon Relat. Elem. 2019, 194, 658–663. [Google Scholar] [CrossRef]
- Saidalimu, I.; Liang, Y.; Niina, K.; Tanagawa, K.; Saito, N.; Shibata, N. Synthesis of Aryl and Heteroaryl Tetrafluoro-λ6-sulfanyl Chlorides from Diaryl Disulfides Using Trichloroisocyanuric Acid and Potassium Fluoride. Org. Chem. Front. 2019, 6, 1157–1161. [Google Scholar] [CrossRef]
- Cui, B.; Kosobokov, M.; Matsuzaki, K.; Tokunaga, E.; Shibata, N. IF5 Affects the Final Stage of the Cl-F Exchange Fluorination in the Synthesis of Pentafluoro-λ6-sulfanyl-pyridines, Pyrimidines and Benzenes with Electron-Withdrawing Substituents. Chem. Commun. 2017, 53, 5997–6000. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Uno, H.; Tokunaga, E.; Shibata, N. Fluorobissulfonylmethyl Iodides: An Efficient Scaffold for Halogen Bonding Catalysts with an sp3-Hybridized Carbon−Iodine Moiety. ACS Catal. 2018, 8, 6601–6605. [Google Scholar] [CrossRef]
- Das, P.; Tokunaga, E.; Akiyama, H.; Doi, H.; Saito, N.; Shibata, N. Synthesis of Fluoro-Functionalized Diaryl-λ3-iodonium Salts and Their Cytotoxicity against Human Lymphoma U937 Cells. Beilstein J. Org. Chem. 2018, 14, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Aakeröy, C.B.; Wijethunga, T.K.; Desper, J.; Đaković, M. Crystal Engineering with Iodoethynylnitrobenzenes: A Group of Highly Effective Halogen-Bond Donors. Cryst. Growth Des. 2015, 15, 3853–3861. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Sato, N. Origin of Attraction in Chalcogen-Nitrogen Interaction of 1,2,5-chalcogenadiazole Dimers. J. Phys. Chem. B 2013, 117, 6849–6855. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, S.; Uchimaru, T.; Wakisaka, A.; Ono, T. Magnitude and Directionality of Halogen Bond of Benzene with C6F5X, C6H5X, and CF3X (X = I, Br, Cl, and F). J. Phys. Chem. A 2016, 120, 7020–7029. [Google Scholar] [CrossRef] [PubMed]
- Pople, J.A.; Head-Gordon, M.; Raghavachari, K. Quadratic Configuration Interaction. A General Technique for Determining Electron Correlation Energies. J. Chem. Phys. 1987, 87, 5968–5975. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic Interaction of a Solute with a Continuum. A Direct Utilization of ab initio Molecular Potentials for the Prevision of Solvent Effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- He, W.; Ge, Y.-C.; Tan, C.-H. Halogen-Bonding-Induced Hydrogen Transfer to C=N Bond with Hantzsch Ester. Org. Lett. 2014, 16, 3244–3247. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.M.; Kniep, F.; Herdtweck, E.; Huber, S.M. Halogen-Bond-Induced Activation of a Carbon–Heteroatom Bond. Angew. Chem. Int. Ed. 2011, 50, 7187–7191. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Kobayashi, Y.; Tsuzuki, S.; Takemoto, Y. Electrophilic Activation of Iodonium Ylides by Halogen-Bond-Donor Catalysis for Cross-Enolate Coupling. Angew. Chem. Int. Ed. 2017, 56, 7653–7657. [Google Scholar] [CrossRef] [PubMed]
- Guha, S.; Sekar, G. Metal-Free Halogen(I) Catalysts for the Oxidation of Aryl(heteroaryl)methanes to Ketones or Esters: Selectivity Control by Halogen Bonding. Chem. Eur. J. 2018, 24, 14171–14182. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Li, Q. Non-additivity of F Substituent in Enhancing the Halogen Bond in C6H5I⋯NCH. Comput. Theor. Chem. 2015, 1070, 21–26. [Google Scholar] [CrossRef]
- Ellington, T.L.; Reves, P.L.; Simms, B.L.; Wilson, J.L.; Watkins, D.L.; Tschumper, G.S.; Hammer, N.I. Quantifying the Effects of Halogen Bonding by Haloaromatic Donors on the Acceptor Pyrimidine. ChemPhysChem 2017, 18, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Estarellas, C.; Frontera, A.; Quiñonero, D.; Deyà, P.M. Radical Cation (C•+−π) and Radical Anion (A•–−π) Interactions with Aromatic Rings: Energetic, Orbitalic and Spin Density Considerations. Phys. Chem. Chem. Phys. 2011, 13, 16698–16705. [Google Scholar] [CrossRef]
- Klapars, A.; Buchwald, S.L. Copper-Catalyzed Halogen Exchange in Aryl Halides: An Aromatic Finkelstein Reaction. J. Am. Chem. Soc. 2002, 124, 14844–14845. [Google Scholar] [CrossRef]
- Joliton, A.; Carreira, E.M. Ir-Catalyzed Preparation of SF5-Substituted Potassium Aryl Trifluoroborates via C–H Borylation and Their Application in the Suzuki–Miyaura Reaction. Org. Lett. 2013, 15, 5147–5149. [Google Scholar] [CrossRef]
- Bowden, R.D.; Comina, P.J.; Greenhall, M.P.; Kariuki, B.M.; Loveday, A.; Philp, D. A New Method for the Synthesis of Aromatic Sulfurpentafluorides and Studies of the Stability of the Sulfurpentafluoride Group in Common Synthetic Transformations. Tetrahedron 2000, 56, 3399–3408. [Google Scholar] [CrossRef]
- Iakobson, G.; Du, J.; Slawin, A.M.Z.; Beier, P. Pyridine-promoted Dediazoniation of Aryldiazonium Tetrafluoroborates: Application to the Synthesis of SF5-substituted Phenylboronic Esters and Iodobenzenes. Beilstein J. Org. Chem. 2015, 11, 1494–1502. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D.J. A new mixing of Hartree–Fock and local density-functional theories. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Becke, A.D.J. Density-functional thermochemistry. III. The role of exact exchange. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 Energy Evaluation by Direct Methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Ransil, B.J. Studies in Molecular Structure. IV. Potential Curve for the Interaction of Two Helium Atoms in Single-Configuration LCAO MO SCF Approximation. J. Chem. Phys. 1961, 34, 2109–2118. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Godbout, N.; Salahub, D.R.; Andzelm, J.; Wimmer, E. Optimization of Gaussian-Type Basis Sets for Local Spin Density Functional Calculations. Part I. Boron through Neon, Optimization Technique and Validation. Can. J. Chem. 1992, 70, 560–571. [Google Scholar] [CrossRef]
- Helgaker, T.; Klopper, W.; Koch, H.; Noga, J. Basis-Set Convergence of Correlated Calculations on Water. J. Chem. Phys. 1997, 106, 9639–9646. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Honda, K.; Uchimaru, T.; Mikami, M.; Tanabe, K. Origin of Attraction and Directionality of the π/π Interaction: Model Chemistry Calculations of Benzene Dimer Interaction. J. Am. Chem. Soc. 2002, 124, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, K.; Fujii, A.; Mikami, M.; Tsuzuki, S. Magnitude and Nature of Interactions in Benzene−X (X = Ethylene and Acetylene) in the Gas Phase: Significantly Different CH/π Interaction of Acetylene As Compared with Those of Ethylene and Methane. J. Phys. Chem. A 2007, 111, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Stone, A.J.; Alderton, M. Distributed Multipole Analysis. Mol. Phys. 1985, 56, 1047–1064. [Google Scholar] [CrossRef]
- Stone, A.J. The Theory of Intermolecular Forces, 2nd ed.; Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- Stone, A.J.; Dullweber, A.; Hodges, M.P.; Popelier, P.L.A.; Wales, D.J. Orient: A Program for Studying Interactions between Molecules, v. 3.2; University of Cambridge: Cambridge, UK, 1995. [Google Scholar]
- Stone, A.J. Distributed Multipole Analysis: Stability for Large Basis Sets. J. Chem. Theory Comput. 2005, 1, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Stone, A.J. Distributed Polarizabilities. Mol. Phys. 1985, 56, 1065–1082. [Google Scholar] [CrossRef]
- Van Duijnen, P.T.; Swart, M. Molecular and Atomic Polarizabilities: Thole’s Model Revisited. J. Phys. Chem. A 1998, 102, 2399–2407. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1a–d are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | E (kcal mol−1) | ||||
|---|---|---|---|---|---|
| Einta | Eesb | Eindc | Eshortd | Ecorre | |
| o-SF5-iodobenzene---pyridine (2a) | −4.88 | −4.48 | −1.32 | 4.43 | −3.52 |
| m-SF5-iodobenzene---pyridine (2b) | −4.27 | −4.20 | −1.20 | 4.40 | −3.27 |
| p-SF5-iodobenzene---pyridine (2c) | −4.28 | −4.02 | −1.15 | 4.02 | −3.13 |
| 3,5-bis-SF5-iodobenzene---pyridine (2d) | −5.21 | −5.60 | −1.65 | 5.54 | −3.49 |
| 3,5-bis-NO2-iodobenzene---pyridine (3) | −4.83 | −5.23 | −1.52 | 4.46 | −2.53 |
| pentafluoro-iodobenzene---pyridine (4) | −5.71 | −5.83 | −1.68 | 5.11 | −3.31 |
| iodobenzene---pyridine (5) | −3.25 | −2.73 | −0.82 | 3.30 | −2.99 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sumii, Y.; Sasaki, K.; Tsuzuki, S.; Shibata, N. Studies of Halogen Bonding Induced by Pentafluorosulfanyl Aryl Iodides: A Potential Group of Halogen Bond Donors in a Rational Drug Design. Molecules 2019, 24, 3610. https://doi.org/10.3390/molecules24193610
Sumii Y, Sasaki K, Tsuzuki S, Shibata N. Studies of Halogen Bonding Induced by Pentafluorosulfanyl Aryl Iodides: A Potential Group of Halogen Bond Donors in a Rational Drug Design. Molecules. 2019; 24(19):3610. https://doi.org/10.3390/molecules24193610
Chicago/Turabian StyleSumii, Yuji, Kenta Sasaki, Seiji Tsuzuki, and Norio Shibata. 2019. "Studies of Halogen Bonding Induced by Pentafluorosulfanyl Aryl Iodides: A Potential Group of Halogen Bond Donors in a Rational Drug Design" Molecules 24, no. 19: 3610. https://doi.org/10.3390/molecules24193610