1. Introduction
Lung cancer is the most commonly diagnosed form of cancer and the primary cause of cancer-related mortality for males worldwide. It is also the second leading cause of cancer-related deaths among women globally [
1]. Lung cancer is aggressive, and its treatment remains a difficult and challenging task for physicians [
2]. Several research studies have indicated that long-term exposure to inhaled carcinogens has the greatest impact on increased risks of lung cancer [
1,
3,
4,
5]. Inhalation of such toxic air pollutants and microorganisms can cause lung injuries and chronic inflammation [
6]. Chronic inflammation has been associated with cancer development. Many proinflammatory mediators, especially cytokines, chemokines, and prostaglandins, have been found to promote cancer proliferation, invasion, angiogenesis, and drug resistance [
7].
A large number of studies have indicated that TNF-α displays a degree of potential in linking the molecules associated with inflammation and cancer. The data obtained from clinical studies have revealed that the expression level of TNF-α in the tumor tissues and serum samples obtained from patients with non-small cell lung cancer increased along with the clinical stage of the tumor [
8,
9]. TNF-α plays an important role in the process by binding itself to the tumor necrosis factor R-1 (TNFR-1). After the binding of TNF-α and TNFR-1, the receptor interacts with TRADD (TNFR1-associated death domain) to initiate the recruitment of receptor-interacting protein 1 (RIP1) and TNFR-associated factor 2 (TRAF2) [
10]. These complex signaling lead to induce cancer cell survival, proliferation, and metastasis via upregulation of antiapoptotic (Survivin, XIAP, Bcl-xl, Bcl-2, and cFLIP), proliferative (cyclin D and cyclin B1), invasive (MMP-9, MT1-MMP, uPA, and uPAR), and angiogenic (VEGF, and COX-2) proteins by activating NF-κB, activator protein-1 (AP-1), and the mitogen-activated protein kinase (MAPKs) signaling pathway [
11,
12]. On the other hand, the binding of TNF-α and TNFR-1 can induce the program cell death that is involved with apoptosis by recruiting TRADD-FADD and caspases-8. In fact, TRADD and caspase-8 complex are assembled over a delayed period of time when compared with TRAF2 and RIP1, which results in a sufficient amount of time needed to activate survival signaling. Therefore, the expression of anti-apoptotic proteins and caspase inhibitors, including Bcl-2, Bcl-xl, xIAP, and cFLIP, would be elevated prior to caspase 8 activation [
13,
14]. Thus, the blockade of TNF-α-induced survival signaling can lead to an increase in the sensitivity of TNF-α-induced cell death. Moreover, many studies have shown that TNF-α expression results in the induction of multiple autophagy markers in breast cancer cells, lung cancer cells, and Ewing’s sarcoma cells [
15]. A novel function of anti-apoptotic proteins, such as cFLIP, survivin, Bcl-2, and Bcl-xl, that serve as autophagy inhibitors, have been reported in various cells [
16,
17]. Downregulation of these antiapoptotic proteins could enhance TNF-α-induced cancer cell death via autophagy and apoptosis. Accordingly, the efficient agents that can suppress TNF-α-induced cancer cell progression could be an important part of an attractive and alternative form of cancer therapy.
Proanthocyanidins, also known as condensed tannins, are a class of polymeric phenolic compounds that consist mainly of catechin, epicatechin, gallocatechin, and epigallocatechin units [
18]. Recently, our previous findings have demonstrated that proanthocyanidin-rich fractions derived from red rice (PRFR) inhibited inflammation in LPS-treated Raw 264.7 cells via suppression of the AP-1, NF-κB, and MAPKs signaling pathways [
19]. Moreover, PRFR reduced human fibrosarcoma, HT1080 cells and breast adenocarcinoma, MDA-MB-231 cells invasion via inhibition of the expression of invasive proteins [
20]. Furthermore, PRFR suppressed cell proliferation in human hepatocellular carcinoma, HepG2 cells via the downregulation of survival proteins and induced cell apoptosis by enhancing active apoptotic proteins [
21].
However, the effect of PRFR on TNF-α-induced cancer progression has not yet been clarified. Therefore, the purpose of this study was to investigate whether PRFR exerts anticancer effects through suppression of the TNF-α-induced expression of the survival and metastasis proteins by inhibiting the MAPKs, NF-κB, and AP-1 signaling pathways in A549 human lung adenocarcinoma cells.
3. Discussion
Proanthocyanidins are oligomers and polymers of flavanol-3-ol which are found in various fruits, vegetables, and cereals. Notably, they are present in foods such as grape seeds, blackberries, and red rice [
18,
24]. Our previous study revealed that PRFR exhibited anti-cancer activities by inducing HepG2 hepatocarcinoma cell apoptosis and inhibiting MDA-MB-231 breast cancer cell invasion [
20,
21]. Despite its various pharmacological activities, the molecular mechanism of PRFR on the anti-tumor effects in A549 lung adenocarcinoma cells has not been elucidated. In this study, we investigated whether PRFR could sensitize TNF-α-induced cell death in lung A549 cancer cells and then act as a potent inhibitor of TNF-α-induced A549 cell metastasis. Molecular mechanisms of this phenomena have been elucidated.
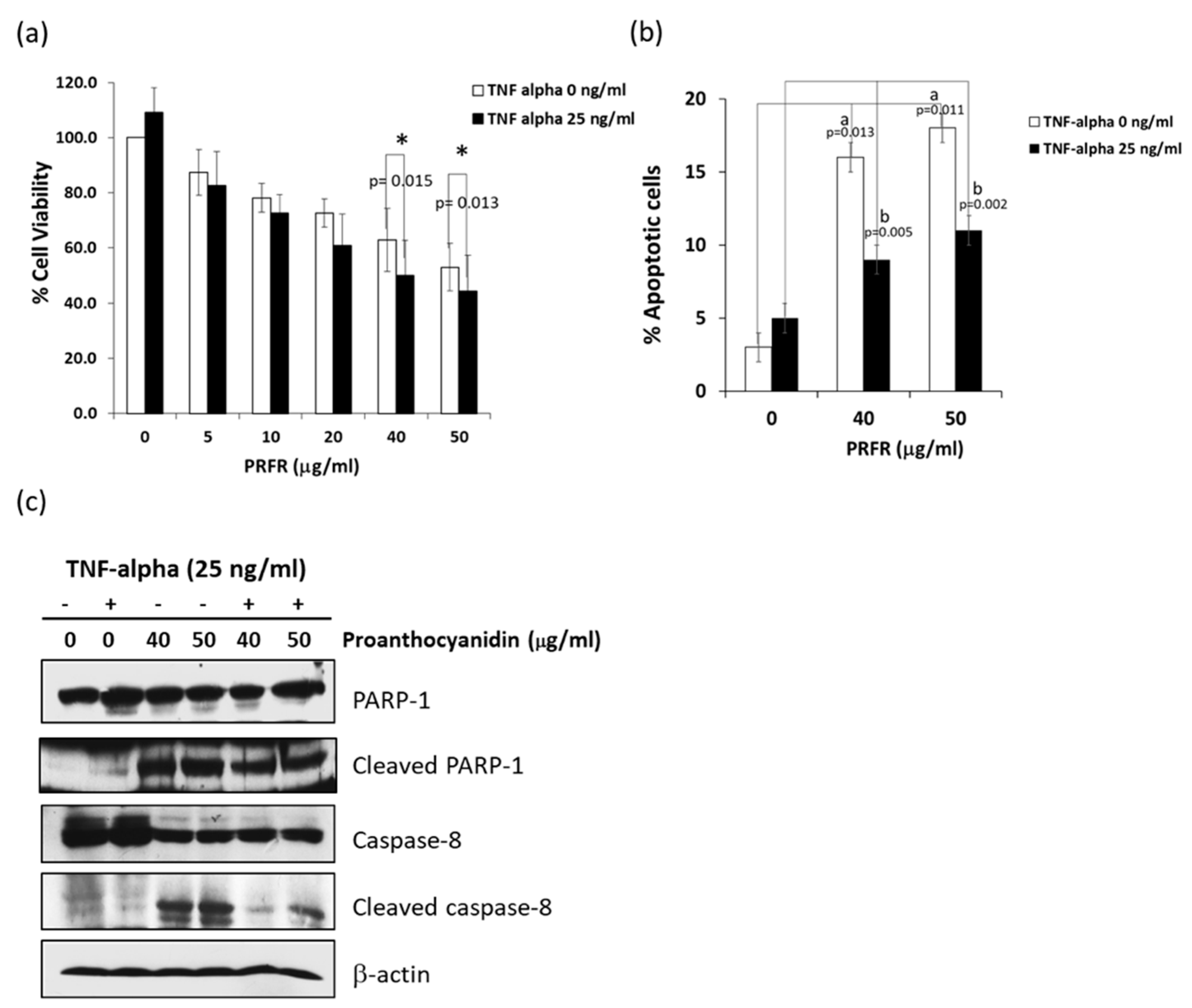
The interaction between TNF-α and TRFR-1 can trigger survival or death signaling pathways. It has been reported that most cancer cells are resistant to TNF-α-induced cell death via increased expressions of survival proteins through the induction transcriptional activity of NF-κB. Thus, a blockade of the activation of the survival signaling pathways may lead to an increase in sensitivity in TNF-α-induced cell death [
12]. This study was the first to report that PRFR sensitized A549 lung adenocarcinoma cells to TNF-α-induced cell death. TNF-αpromotes cancer cell death by induction apoptosis, necroptosis, and autophagy depending on the condition of the cells. TNF-α induced cell apoptosis via the intrinsic and extrinsic pathways. The results of this study demonstrate that PRFR alone enhanced A549 cell apoptosis with increased caspase-8 activation while inducing PARP cleavage. However, a combination of PRFR and TNF-α induced A549 to apoptosis to a lesser degree than PRFR alone and also revealed a partial effect on caspase-8 activation and the level of the cleaved PARP. This would suggest that apoptosis is more than a mechanism for PRFR to enhance TNF-α-induced A549 cell death.
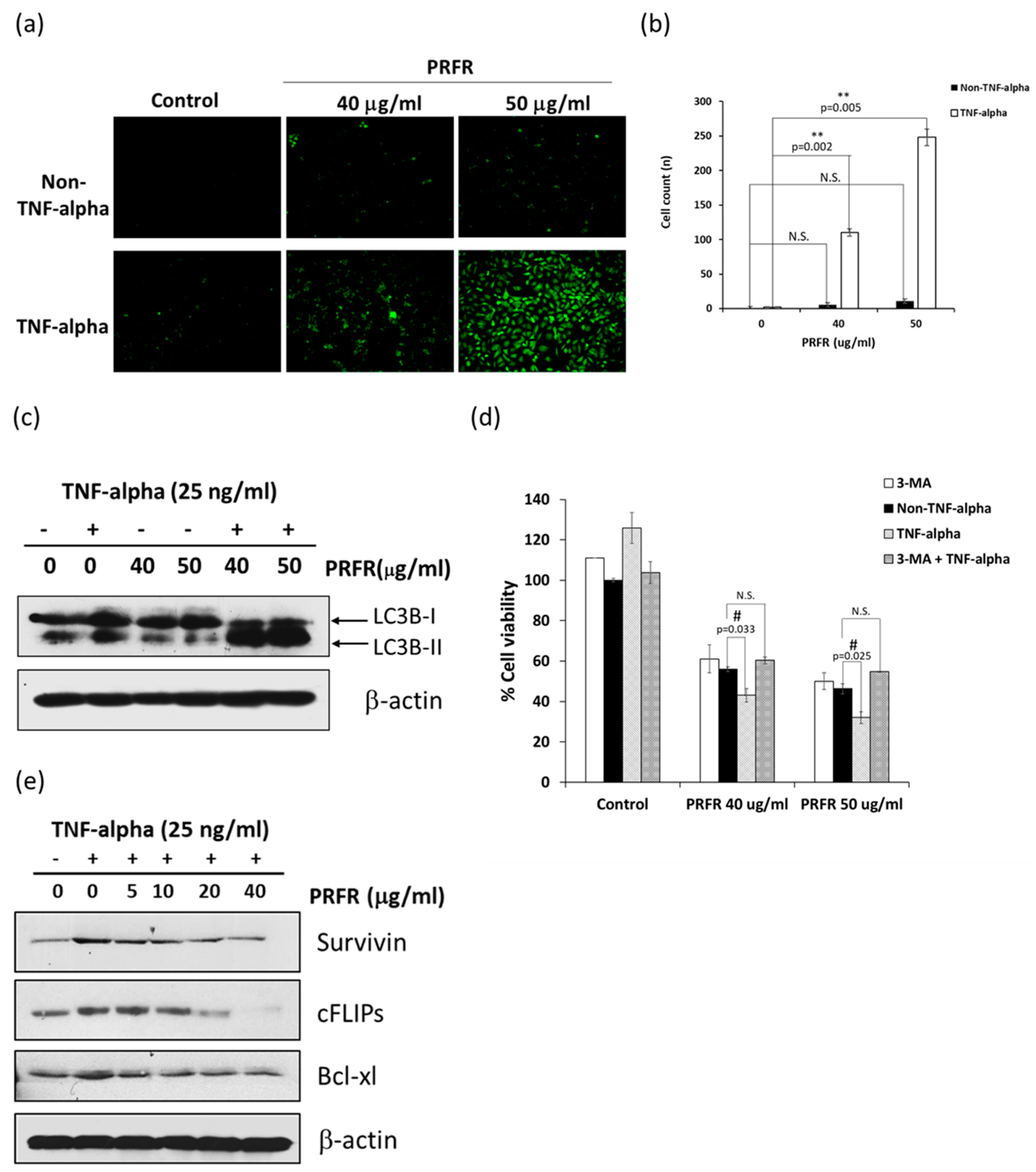
Autophagy has been extensively reported to play a critical role in the control of cell proliferation, differentiation, and cell death. Autophagy is a highly regulated and fundamental cellular homeostatic process, in which cytoplasmic material is delivered and organelles convert to lysosomes via double membrane vesicles called autophagosomes for degradation. Autophagy is activated in response to various forms of cellular stress, including starvation, hypoxia, radiation, and inflammation [
25,
26]. Many studies have shown that TNF-α-induced autophagic cell death occurs in various cancer cell types including breast cancer, hepatoma and ovarian cancer [
27,
28]. Therefore, autophagy is considered a potential pathway in the treatment of cancer. Many natural drug molecules, such as curcumin, celastrol, and bufalin, play important roles in tumor inhibition by inducing autophagy. Thus, PRFR was examined whether it can enhance TNF-α-induced A549 cell death via autophagy cell death. Co-treatment of PRFR and TNF-α led A549 cells to autophagy by accumulating autophagosomes and upregulating the expression of LC3B-II proteins. Whereas, PRFR alone did not induce autophagy, LC3s proteins were found to be a structural protein of autophagosome membranes. The conversion of a soluble form of LC3B-I to LC3B-II is often used to demonstrate active autophagy. To confirm that autophagy is a major process of PRFR in the enhancement of TNF-α induced cell death by using 3-MA as an autophagy inhibitor. The obtained results indicate that using 3-MA could reverse the enhancement effect of PRFR on TNF-α-induced cell death, which would indicate that the way in which PRFR enhanced the cytotoxicity effect of TNF-α was due to autophagy cell death. Recent findings have revealed a novel function of anti-apoptotic proteins, such as FLIP, survivin, Bcl-2, and Bcl-xl, as negative regulators of autophagy. FLIP has been shown to inhibit LC3 lipidation by competitive interaction with ATG3, which in turn blocks autophagy [
16]. Moreover, Zhu J., et al. have shown that the inhibition of survivin through the use of siRNA enhanced autophagy by upregulating Beclin-1 [
29]. Therefore, in order to explain the mechanism by which PRFR sensitizes TNF-α-induced autophagy, the modulatory effect of PRFR on TNF-α induced FLIP, Bcl-xl, and survivin expression levels was examined. It was found that the levels of FLIP, Bcl-xl and survivin were reduced by PRFR. Together, these results suggest that the manner in which PRFR enhanced TNF-α induced cell death was at least in part accomplished by down regulating FLIP, Bcl-xl and survivin, which then led to autophagic cell death.
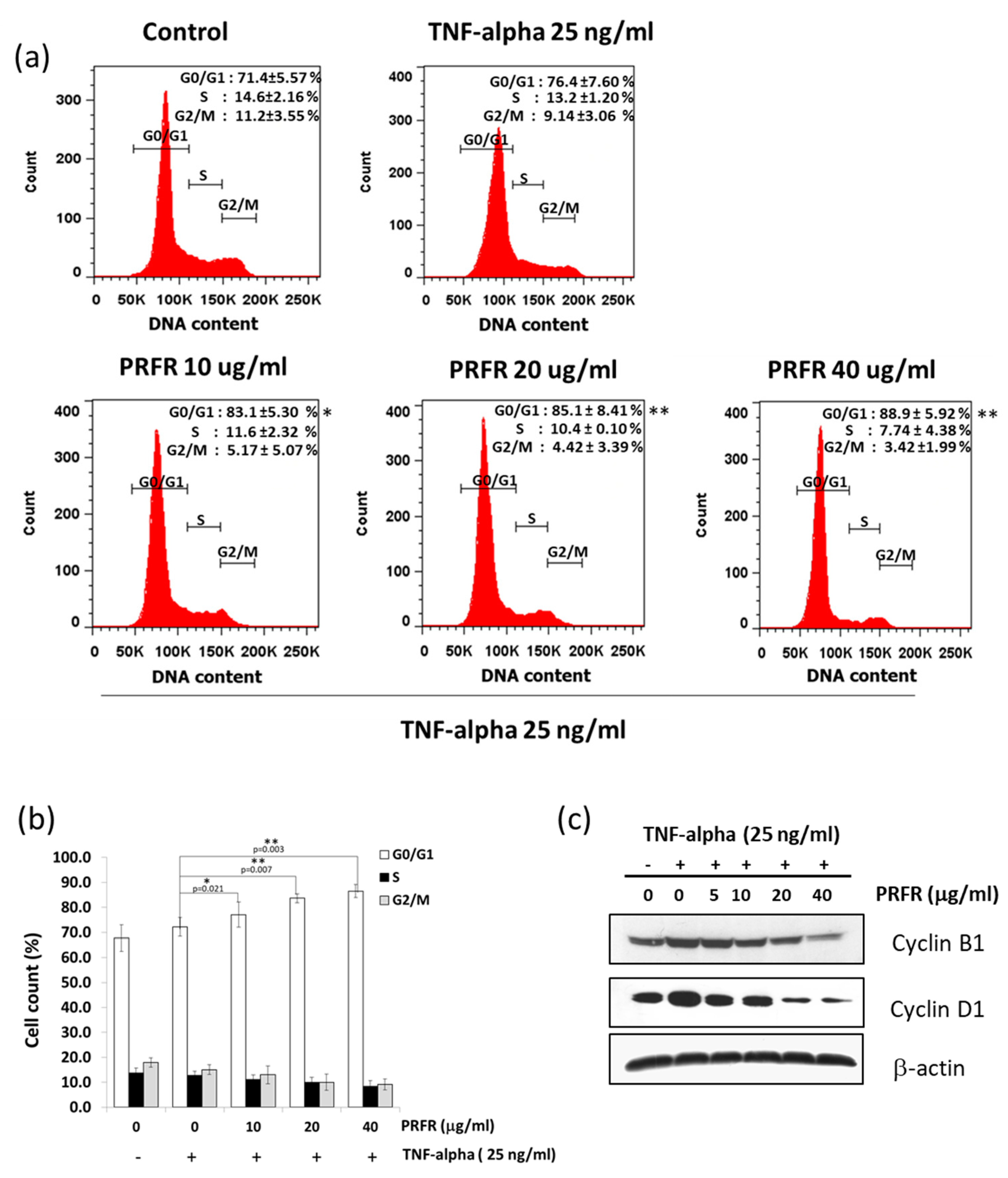
Moreover, the results of this study indicate that PRFR can suppress cell proliferation by blocking cell cycle progression in the G1 phase. TNF-α is known to stimulate transcriptions of Cyclin D1, Cyclin E, and Cyclin B1 in order to accelerate the progression of the cell cycle. Cyclin D1 is a key regulator of the G1 checkpoint control [
30]. This finding is consistent with our observation that PRFR suppressed TNF-α-induced expression of cyclin D1. This result suggests that PRFR reduced TNF-α induced cell proliferation by inhibiting the expression of cyclin proteins.
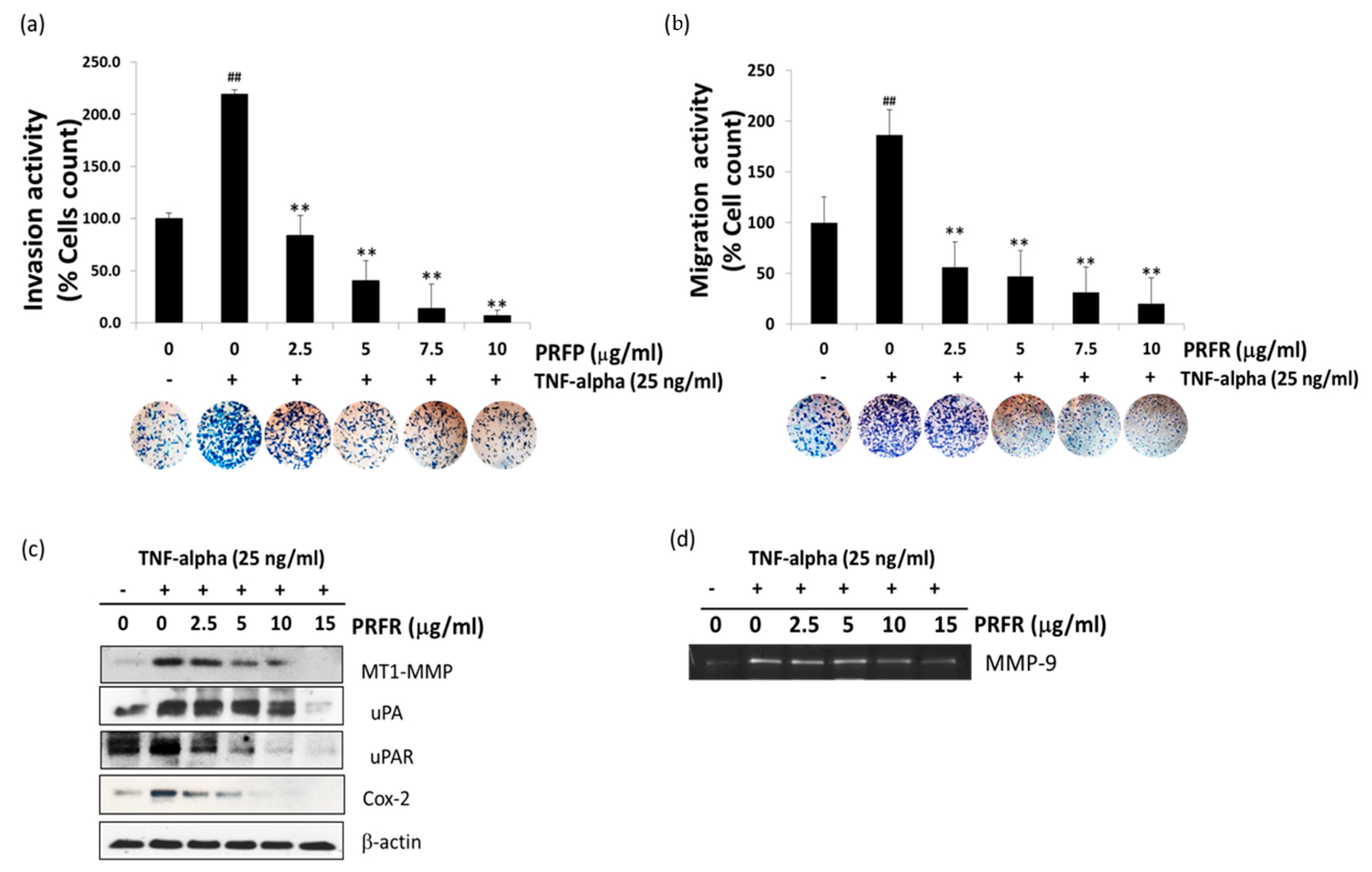
The degradation of ECM and the components of the basement membrane through which proteases are the key steps of cancer cell invasion and metastasis. Of these proteases, MMPs such as MMP-9, MT1-MMP, and uPA are thought to play an important role in cancer invasion. Furthermore, COX-2 has been implicated in metastasis, and its overexpression can enhance cellular invasion, proliferation, and induce angiogenesis [
31,
32,
33]. Previous observations have indicated that TNF-α is an inducer for the invasion and metastasis of A549 cells. These results clearly demonstrate that PRFR inhibits the TNF-α-induced invasion and migration of A549. Moreover, PRER reduced the levels of TNF-α-induced expression of invasive genes, including MMP-9, MT1-MMP, uPA, uPAR, and Cox-2, in the A549 cells.
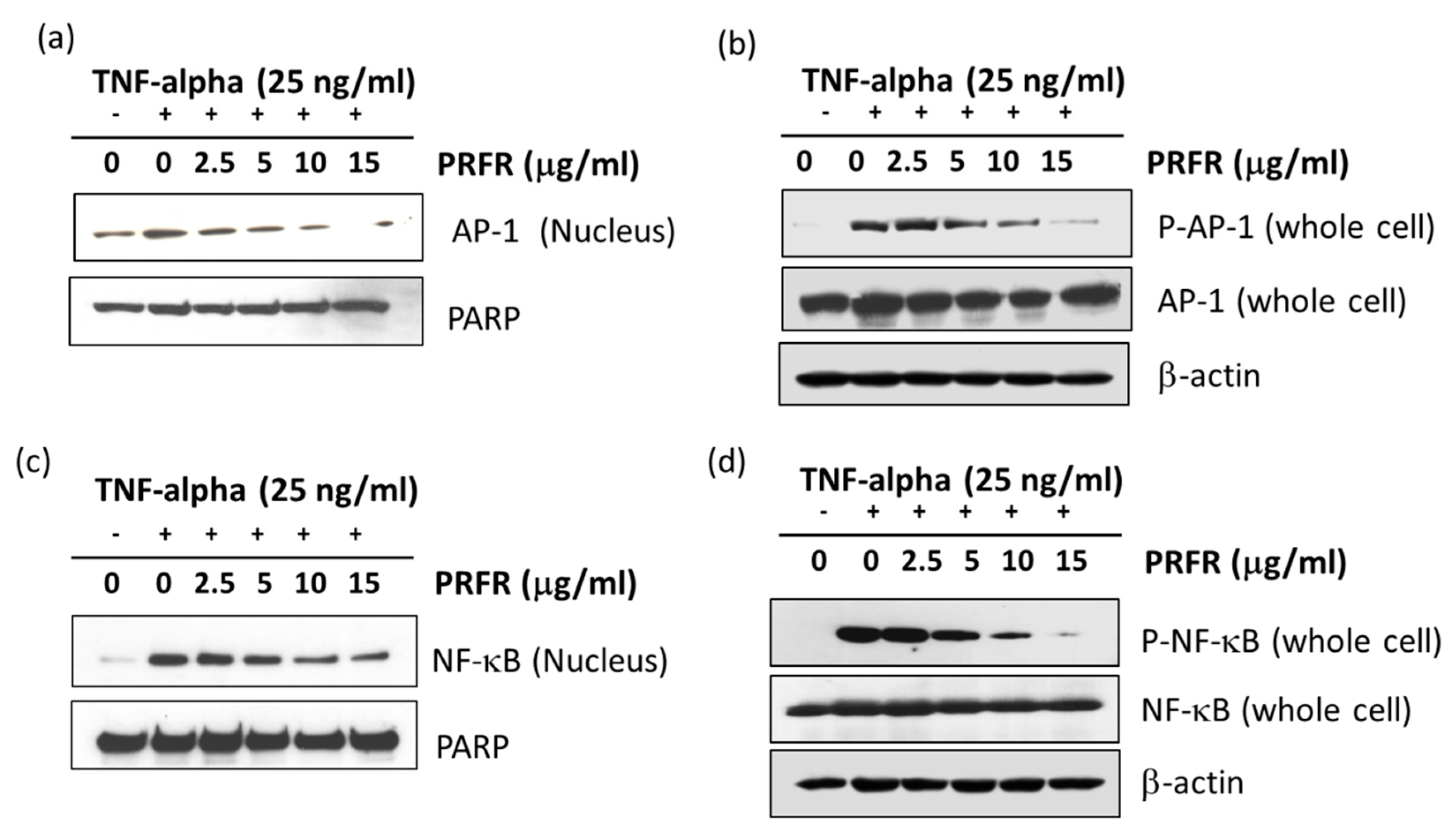
NF-κB and AP-1 are major key players in TNF-α-mediated tumor progression. NF-κB regulates the expression of the survival gene products cIAP, Bcl-2, Bcl-xl, and FLIP, along with the proliferation of gene products cyclin B1 and cyclin D1, and the invasion of gene products uPA, COX-2, MMP-9, and MT1-MMP, which are known to be induced by TNF-α [
31,
34,
35]. Furthermore, it has been reported that TNF-α could induce autophagy in cancer cells when NF-κB signaling is inhibited [
36,
37]. A common form of NF-κB is a heterodimer consisting of p50/p65. NF-κB is normally retained in the cytoplasm through interaction with its inhibitor IκB. Upon TNF-α stimulation, IκB-α is catalyzed for phosphorylation by IκB kinase (IKK) leading to IκB-α degradation and allowing for the nuclear translocation of NF-κB, which promotes the transcription of the corresponding genes. Therefore, we have determined the activity of PRFR on TNF-α can induce the degradation of IκB-α and the translocation of NF-κB activity. Our results demonstrated that PRFR prevented degradation of IκB-α and reduced NF-κB activity by inhibiting TNF-α-induced p65 phosphorylation and translocation to the nucleus of the cells. AP-1 has been implicated in regulating cancer cell survival and proliferation. AP-1 also controls the gene expression values of MMP-9, MT1-MMP, Cox-2, uPA, and uPAR. Here, the activation of AP-1 was investigated by observing the phosphorylation and translocation of c-Jun in TNF-α treated cells. In this study, PRFR inhibited TNF-α induced c-Jun phosphorylation and translocation to the nucleus of the A549 cells. This result was in accordance with the findings of an investigation conducted by Qiao Y et al., which also demonstrated that the suppression of AP-1 signaling can potentiate TNFα-induced cell death and inhibit cancer cell invasion [
34]. Based on the above-mentioned results, we suggest that PRFR could decrease the level of expression of survival and metastasis proteins by the inhibition of AP-1 and NF-κB activation and are also in agreement with inhibition of AP-1 and NF-κB by epigallocatechin gallate reduced cancer cells survival and metastasis [
38,
39].
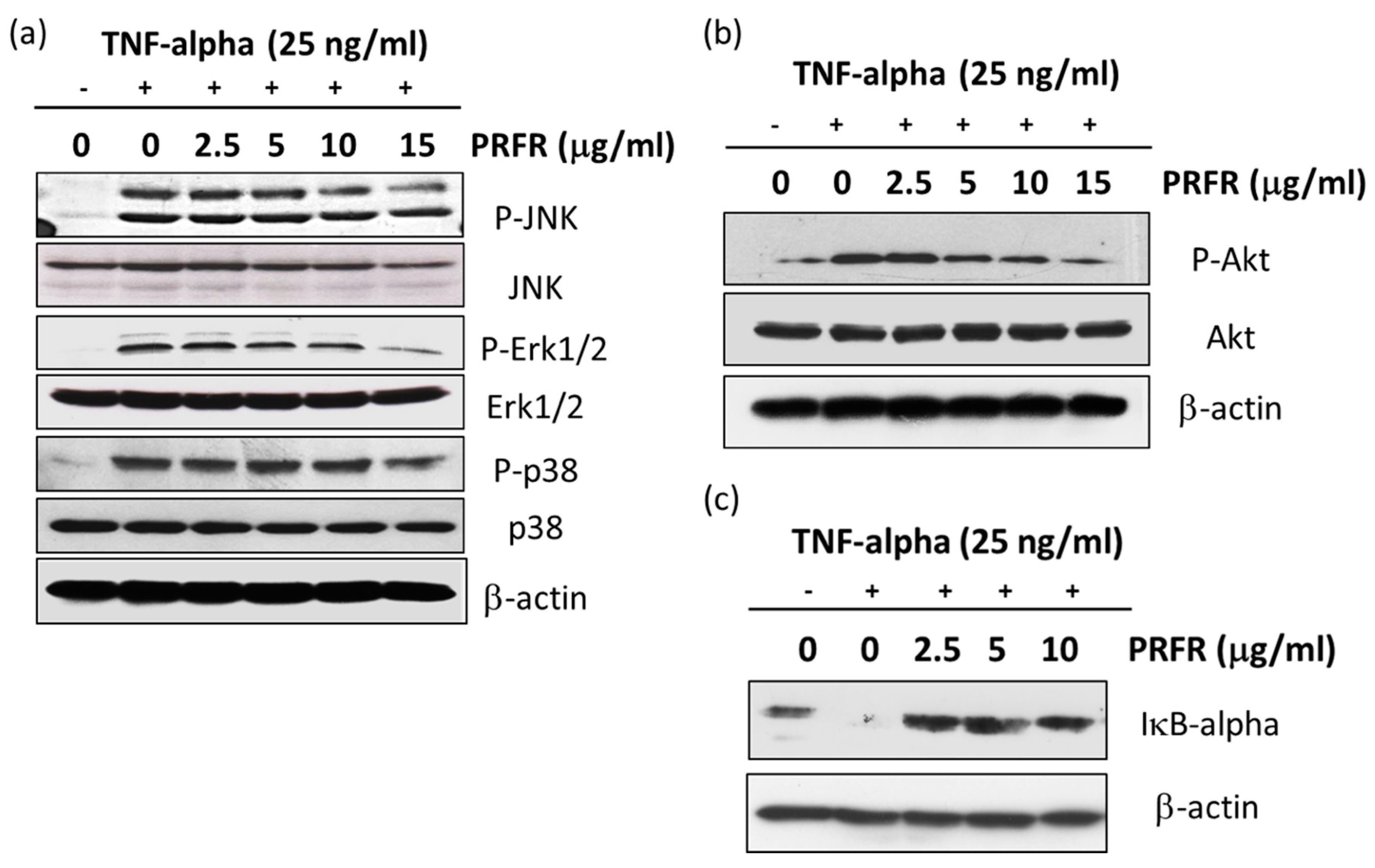
It is accepted that the activation of the MAPKs or Akt signaling pathways is important for regulating survival and metastasis in a variety of cancer cells. TNF-α is bound to the TNF-α receptor-1 which induces NF-κB activation by activating the MAPKs, Akt, and IKK signaling pathways. Moreover, the activation of MAPKs and Akt are important for regulating AP-1 activity. MAPKs are known to be serine/threonine kinase and are composed of several subgroups, such as ERK1/2, JNK, and p38 [
35]. It is generally demonstrated that MAPKs signaling pathways regulate metastasis and survival in a variety of cancer cells. Accumulated evidence indicates that the Akt and MAPKs signaling pathways are involved with autophagy. The AKT/mTOR signaling pathway is one of the survival regulatory pathways in both normal and cancer cells, and it can negatively regulate autophagy [
40]. Therefore, the experiments were performed to determine whether PRFR regulates TNF-α in order to stimulate the activity of MAPKs and Akt. Our results show that PRFR prevented the phosphorylation of p38, ERK, JNK, and Akt. These results are consistent with those of previous reports which have found that using the inhibitors of the PI3K/Akt and MAPK signaling pathways causes cell death and is associated with autophagy, apoptosis and a reduction in the invasive properties of cancer cells.
4. Materials and Methods
4.1. Chemicals and Reagents
Dulbecco’s Modified Eagle Medium (DMEM), trypsin and penicillin-streptomycin were supplied from Gibco (Grand Island, NY, USA). Fetal bovine serum (FBS), RIPA buffer, protease inhibitors and Coomassie Plus™ Protein Assay Reagent were obtained from Thermo Scientific Company (Waltham, MA, USA). Guava Cell Nexin Reagent was purchased from Guava Technologies (Darmstadt, Germany). Nitrocellulose membrane and ECL reagent were supplied from GE Healthcare (Little Chalfont, UK). Gelatin, propidium iodide (PI) and 3-Methyladenine (3-MA) were obtained from Sigma (St. Louis, MO, USA). Antibodies specific to COX-2, and cyclin -D1 were purchased from Millipore (Darmstadt, Germany). Antibodies specific to β-actin, uPA, urokinase-type plasminogen activator receptor (uPAR), poly (ADP-ribose) polymerase (PARP), and p65 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies for the detection of ERK1/2, p38, JNK, c-Jun, and p65 were purchased from Cell Signaling Technology (Danvers, MA, USA). Matrigel was purchased from Becton Dickinson (Bedford, MA, USA).
4.2. Preparation of Proanthocyanidin-Rich Fraction from Red Rice Extract
Whole grains of red rice (Oryza sativa L.) collected from Doi Saket District (Chiang Mai, Thailand) were dehulled and polished in order to obtain the rice germ and bran using a rice de-husker and a rice milling machine (Kinetic (Hubei) Energy Equipment Engineering Co., Ltd., Wuhan, Hubei, China). A voucher specimen was certified by the herbarium at the Flora of Thailand, Faculty of Pharmacy, Chiang Mai University (voucher specimen no. 023148).
Proanthocyanidin-rich fraction (PRFR) was prepared by following the previously reported protocol [
20]. Briefly, 440 g red rice bran were soaked in 50% ethanol for 24 h. After that, the mixture was filtered to separate the ethanolic fractions. The ethanolic fractions were evaporated and partitioned with saturated butanol. The saturated butanol fractions were collected and evaporated to obtain the medium polar fractions. Next, the PRFR was prepared form the medium polar fractions by using Sephadex LH20 (GE Healthcare) chromatography (GE Healthcare Ltd., Little Chalfont, UK). The medium polar fractions (3.5 g) were dissolved in methanol and loaded onto a Sephadex LH-20 column. The fractions were sequentially eluted with solutions of 70% methanol, 30% methanol, and 70% acetone, respectively. Total contents of proanthocyanidins in each fraction were determined by vanillin assay. The fractions containing high concentrations of proanthocyanidins were pooled together and freeze-dried in order to obtain PRFR powder. The total amount of proanthocyanidins in the PRFR was 177.22 ± 16.66 mg catechin/g extract.
4.3. Cell Cultures
A549 lung adenocarcinoma cells were supplied by ATCC. The cells were cultured in DMEM supplemented with 100 U/mL penicillin, and 100 μg/mL streptomycin plus 10% FBS. The cultures were maintained in a humidified incubator with an atmosphere comprised of 95% air and 5% CO2 at 37 °C. For the PRFR treatment, PRFR was dissolved in DMSO and diluted with culture medium, for which the final concentration of DMSO was less than 0.1% (v/v).
4.4. Cell Viability Assay
The cell viability assay of PRFR against A549 lung adenocarcinoma cells was evaluated using trypan blue staining. Briefly, 2 × 104 cells/well were seeded in a 24-well plate and incubated at 37 °C, 5% CO2 for 24 h in DMEM containing 10% FBS. After that, the cells were treated with or without various concentrations (0–200 μg/mL) of PRFR in DMEM containing 10% FBS for 24 h. At the end of the treatment, the percent of cell viability was also determined from counts of the cells suspended in the medium and counts of those cells removed from the plates by trypsinization. Equal parts of 0.4% trypan blue dye were added to the cell suspension in order to obtain a 1 to 2 ratio. The cell viability in each well was determined using Trypan blue dye and the values were compared with the controls.
4.5. Cell Cycle Arrest Assay
A549 cells were incubated with or without various concentrations of the proanthocyanidin-rich fraction (0–50 µg/mL) for 24 h. Then, the cell suspension was prepared on ice and stained with propidium iodide (PI) for 30 min in the dark. Cells were washed with cold PBS and resuspended in 500 μL. For cell cycle analysis, 1 × 104 events were recorded and then analyzed with the BD FACScanTM flow cytometer (BD Biosciences, San Jose, CA, USA).
4.6. Apoptosis Assay
A549 cells were incubated with or without various concentrations of PRFR (0–50 µg/mL) for 24 h. The cell suspension was then prepared and stained with annexin V and 7-amino actinomycin D (Guava Cell Nexin Reagent; Guava Technologies) for 20 min according to the Guava Nexin Assay protocol. Apoptosis was determined on a Guava PCA Instrument using Guava® ViacountTM Software (Merck Ltd., Darmstadt, Germany).
4.7. Extraction of Nuclear and Whole-Cell Lysate
Whole-cell extraction was done to determine the expression levels of the invasive, apoptotic and survival proteins in the A549 cells. The cells were pretreated with various concentrations of PRFR for 4 h and treated with 25 ng/mL of TNF-α for 24 in DMEM medium to determine the levels of uPA, uPAR, COX-2, Survivin, cFLIPs, cyclin D, LC3B, caspase-8, and PARP-1 proteins. The levels of the MAPKs and Akt pathway proteins were determined from the cells treated with PRFR. After that, the cells were treated with TNF-α (25 ng/mL) for 15 min. The treated cells were then extracted using a RIPA lysis buffer containing protease inhibitors (1 mM PMSF, 10 μg/mL leupeptin, 10 μg/mL aprotinin) for 20 min on ice. The insoluble matter was removed by centrifugation at 12,000 rpm for 15 min at 4 °C, and the supernatant fraction (whole cell lysate) was collected and protein concentration was determined using Bradford protein assay.
For the preparation of the nuclear extract fractions, after the A549 cells were treated with PRFR (0–15 μg/mL), TNF-α (25 ng/mL) was added to the cells and they were incubated for 1 h at 37 °C. The treated cells were then collected and the cell pellets were suspended with 50 μL of lysis buffer (10 mM HEPES, pH 7.9, 10 mM KC1, mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl -fluoride, 0.1 μg/mL leupeptin, 1 μg/mL aprotinin). The cells were allowed to swell on ice for 20 min, after which, 15 μL of 10% of Nonidet P-40 was added. The tubes were agitated on a vortex and centrifuged at 12,000 rpm for 5 min. The supernatant was collected and was representative of the cytoplasm extract. The nuclear pellets were suspended in ice-cold nuclear extraction buffer (20 mM HEPES, pH 7.9, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, 1 mM phenylmethylsulfonylfluoride, 2.0 μg/mL leupeptin, 2.0 μg/mL aprotinin) with an intermittent vortex for 30 min. The nuclear extract was centrifuged at 12,000 rpm for 10 min, and the supernatant was collected and used to determine the resulting yield of nuclear proteins.
4.8. Western Blotting Analysis
The whole cell lysate or nuclear extractions were subjected to 10–12% SDS-PAGE. The proteins were transferred onto nitrocellulose membranes. The membranes were blocked with 5% non-fat dried milk protein in 0.5% TBS-tween. Thereafter, the membranes were further incubated overnight with the desired primary antibody at 4 °C followed by incubation with horseradish peroxidase conjugated secondary antibody. Bound antibodies were detected using the chemiluminescent detection system and then exposed to the X-ray film (GE Healthcare Ltd., Little Chalfont, U.K.). Equal values of protein loading were confirmed as each membrane was stripped and re-probed with an anti-β-actin antibody.
4.9. Monodansylcadaverine Staining
The treated A549 cells were stained with 0.05 mM Monodansylcadaverin (MDC) in PBS for 30 min at 37 °C. The cells were washed three times with PBS to remove excess MDC. The visualization step employed a Carl Zeiss Microscopy GmbH (Carl Zeiss AG, Jena, Germany) with an excitation wavelength of 460–500 nm and an emission wavelength of 512–542 nm.
4.10. Statistical Analysis
All data are presented as mean ± standard deviation (S.D.) values. Statistical analysis was analyzed with Prism version 6.0 software GmbH (GraphPad Software, Inc. , San Diego, CA, USA) using one-way ANOVA with Dunnett’s test. Statistical significance was determined at * p < 0.05, ** p < 0.01, *** p < 0.001, or **** p < 0.0001.



 and
and 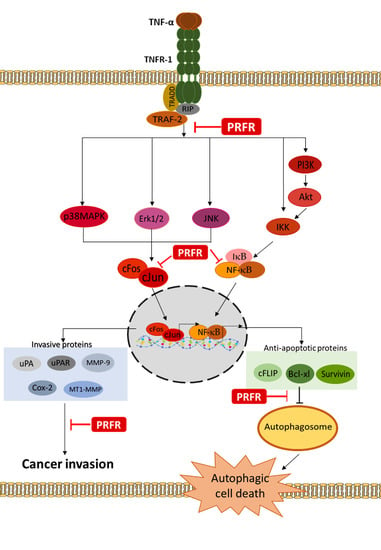
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





