
Palladium Supported on Porous Chitosan–Graphene Oxide Aerogels as Highly Efficient Catalysts for Hydrogen Generation from Formate



Abstract
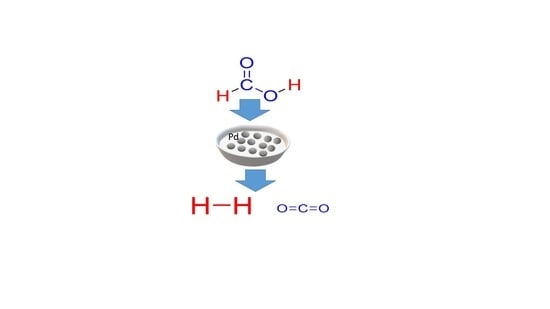
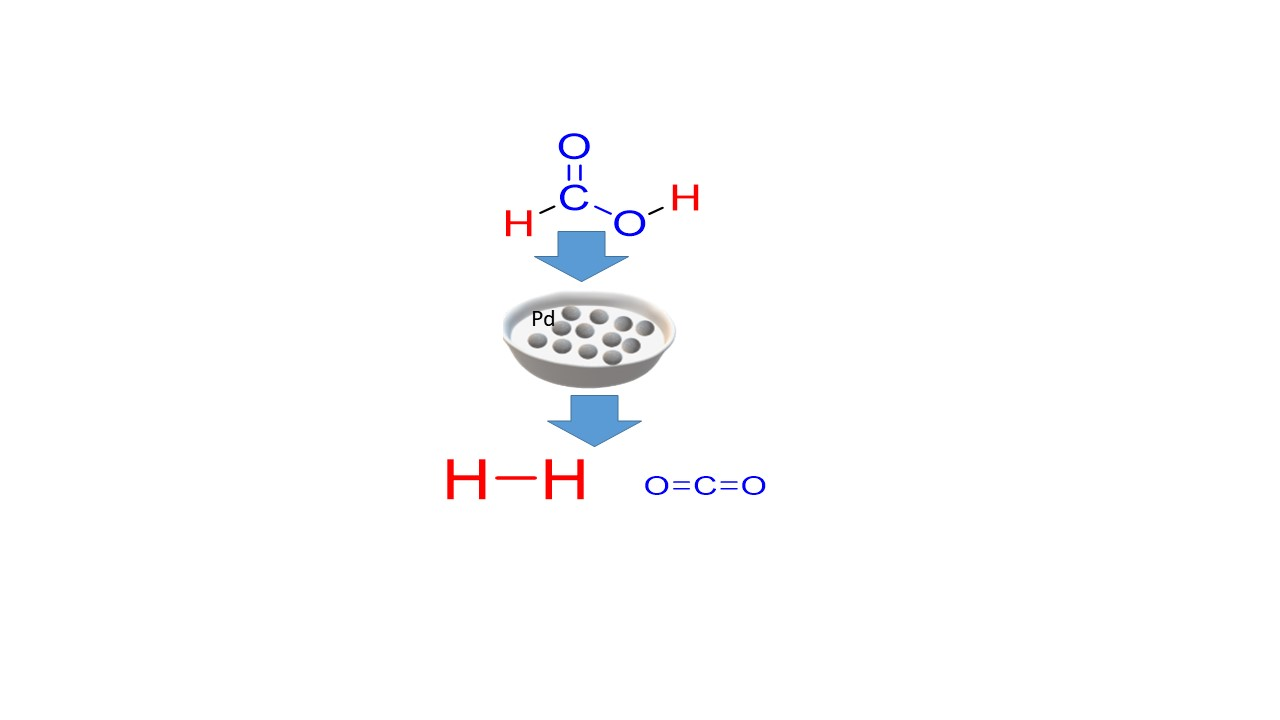
:
1. Introduction
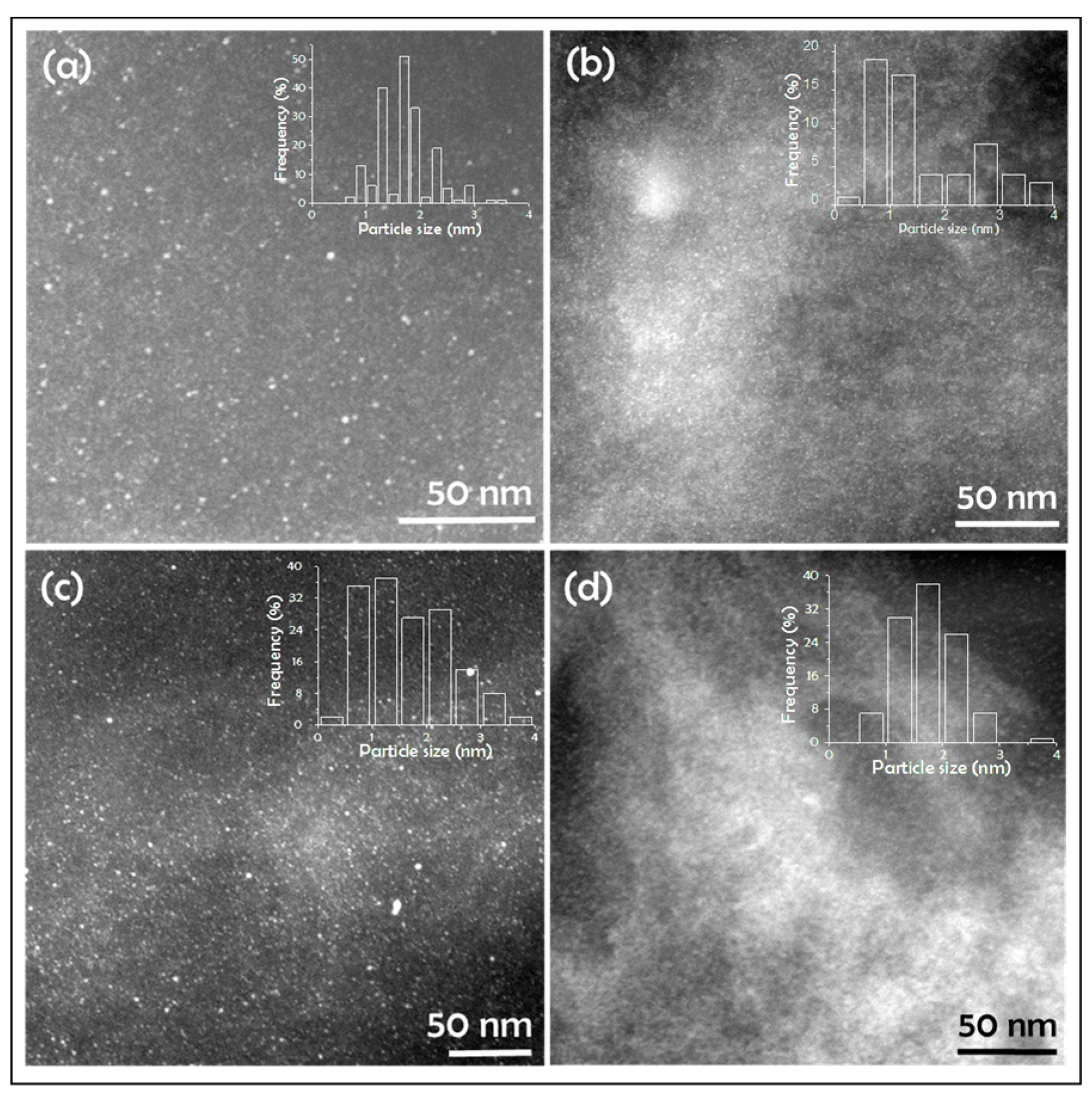
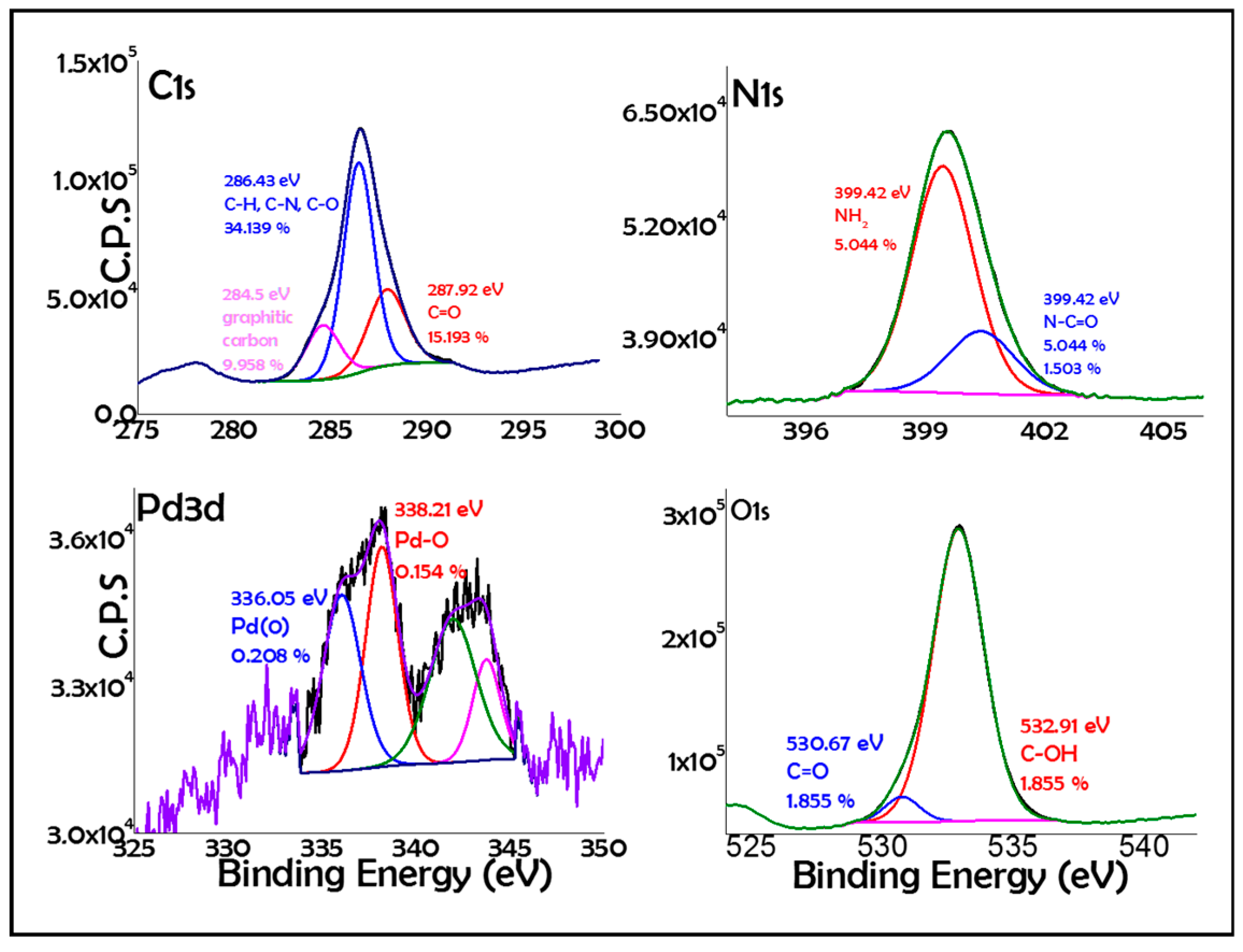
2. Results
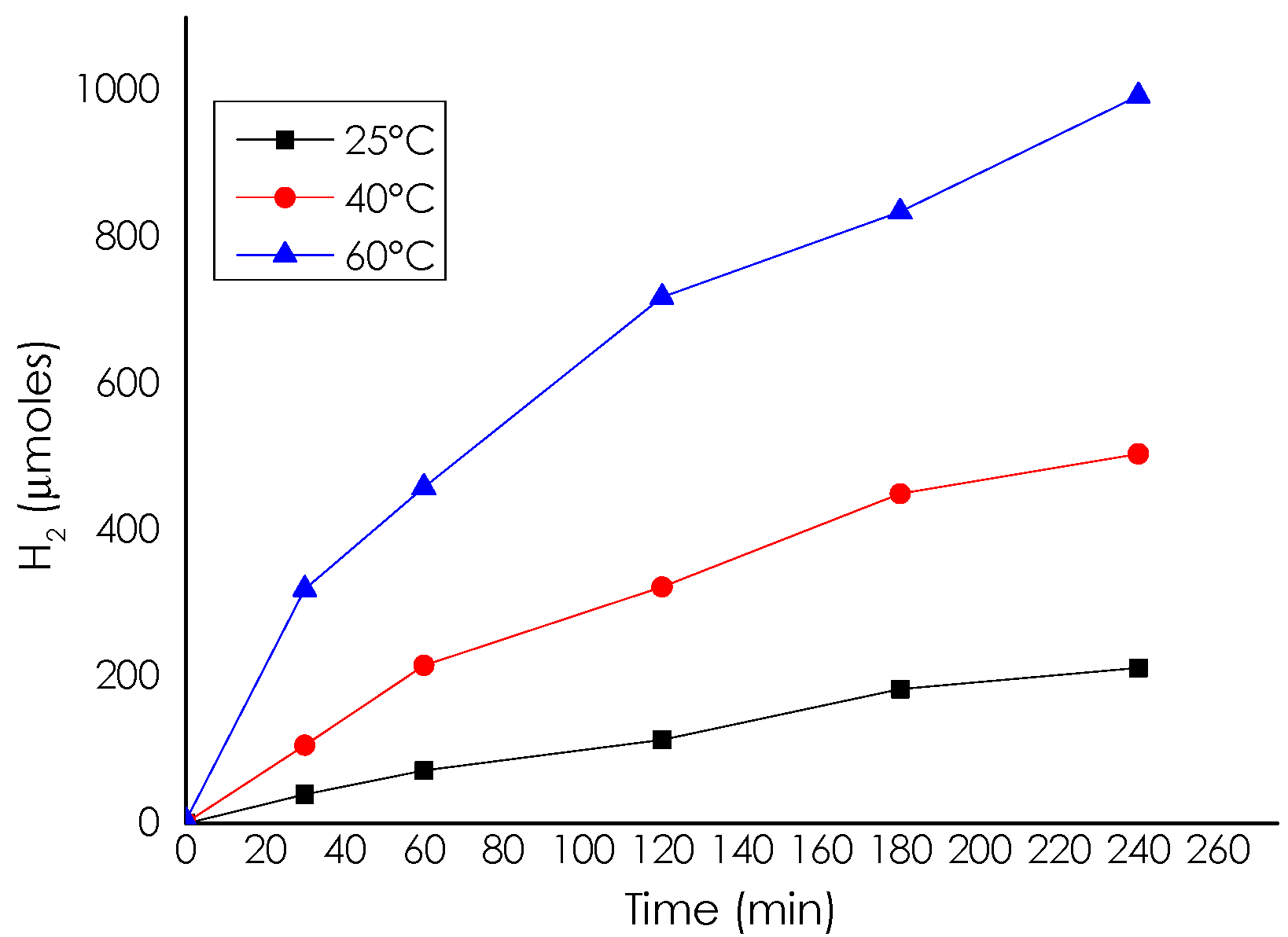
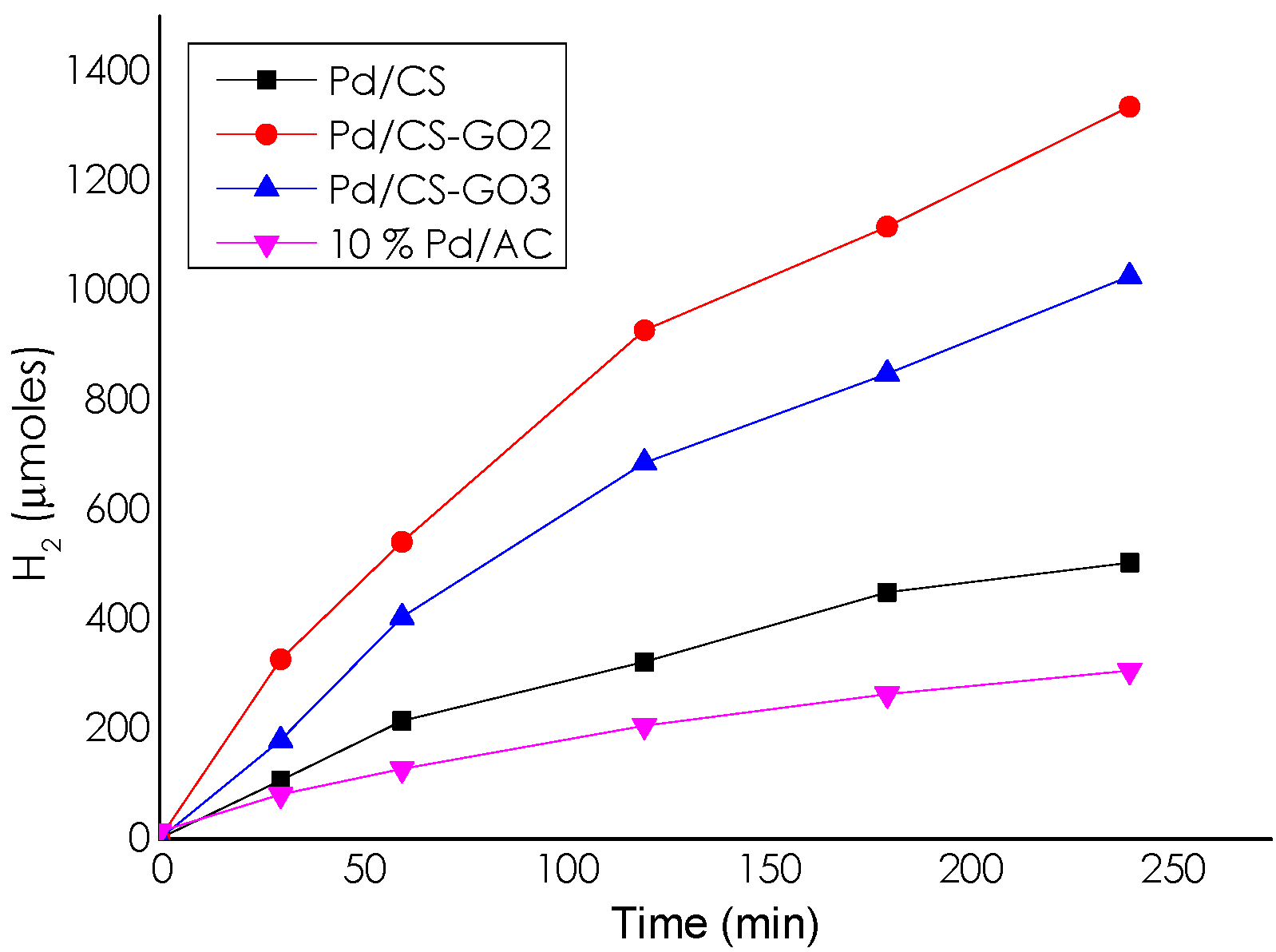
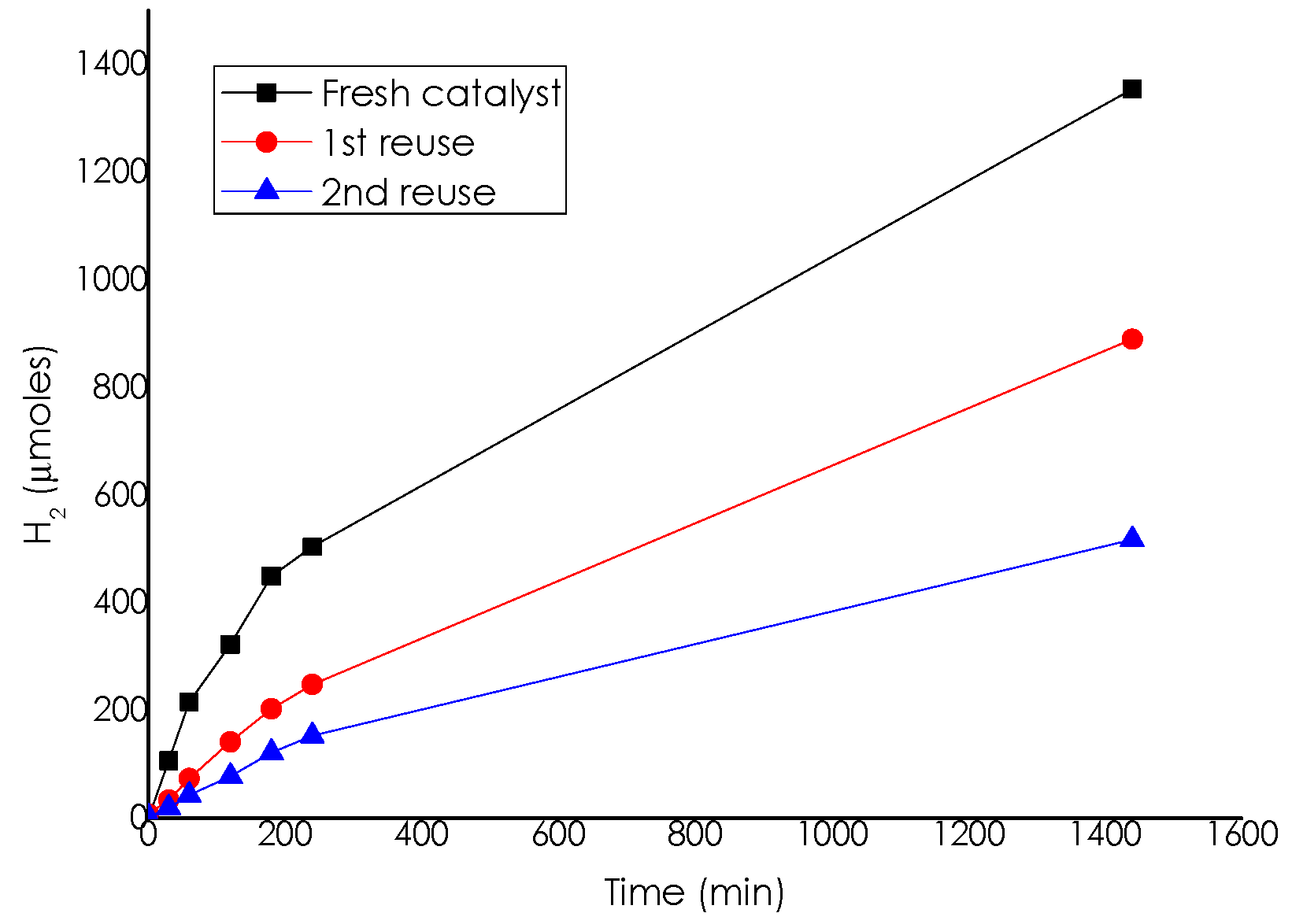
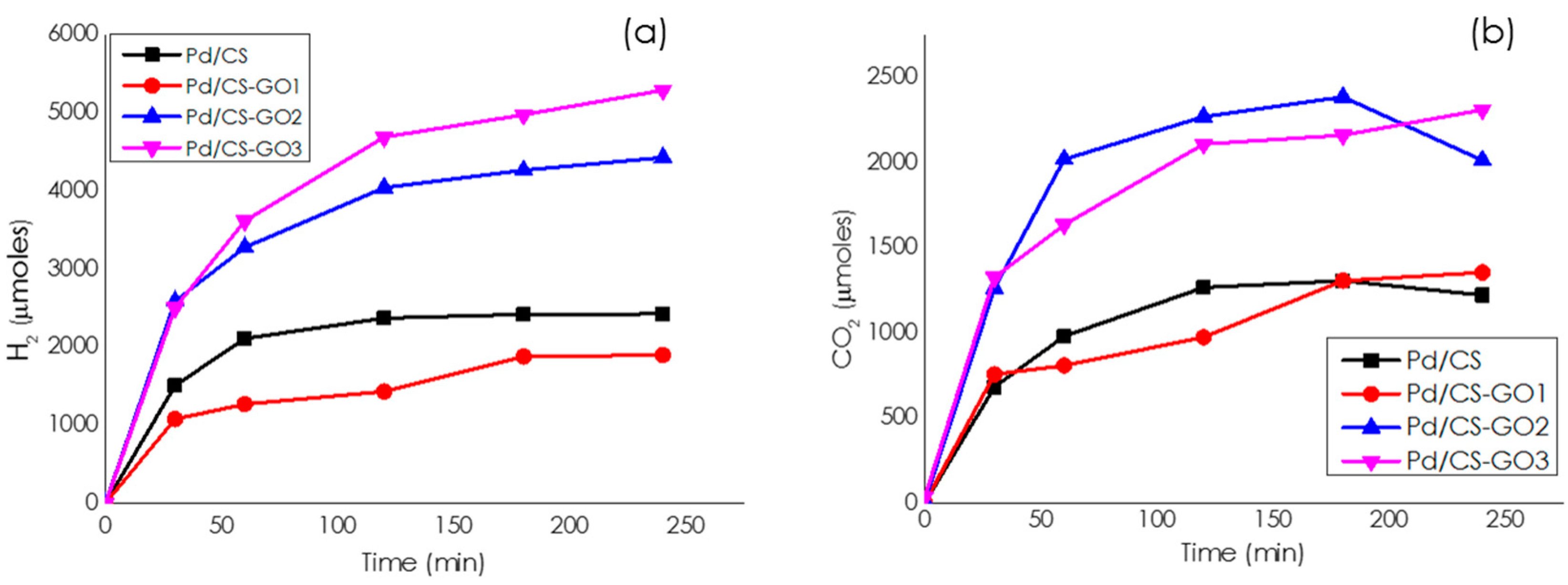
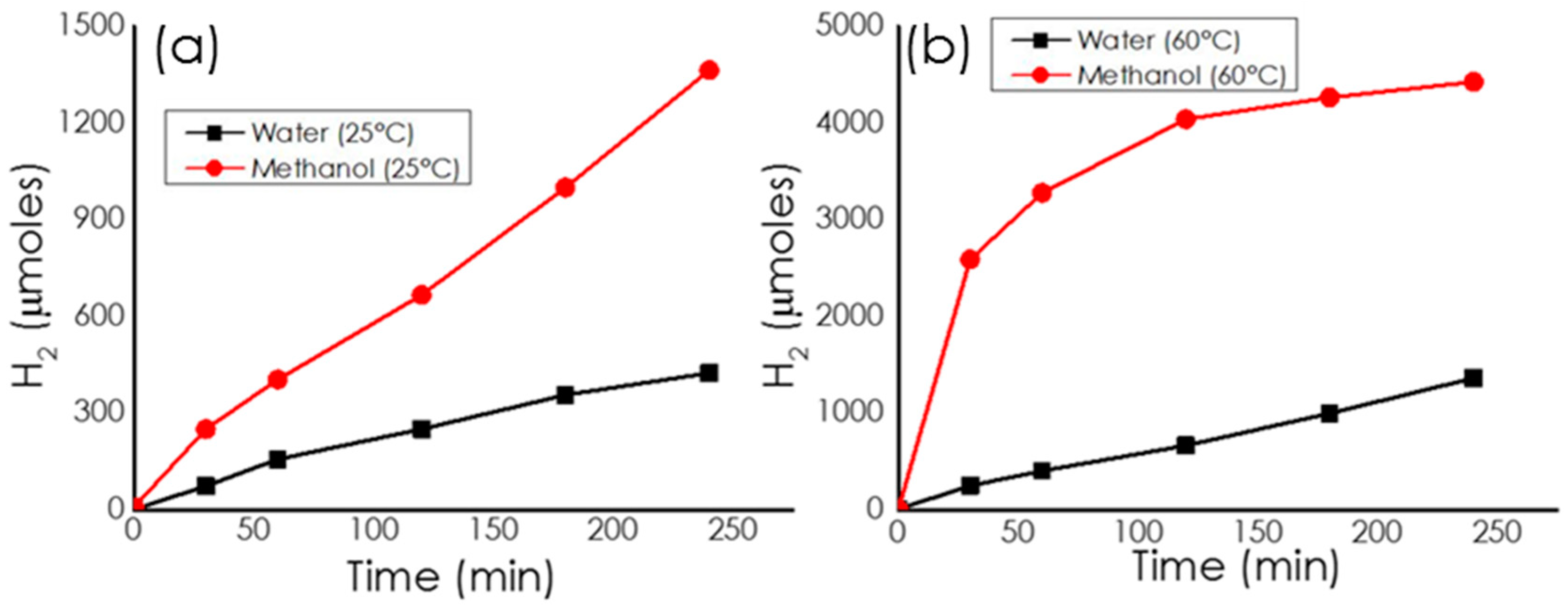
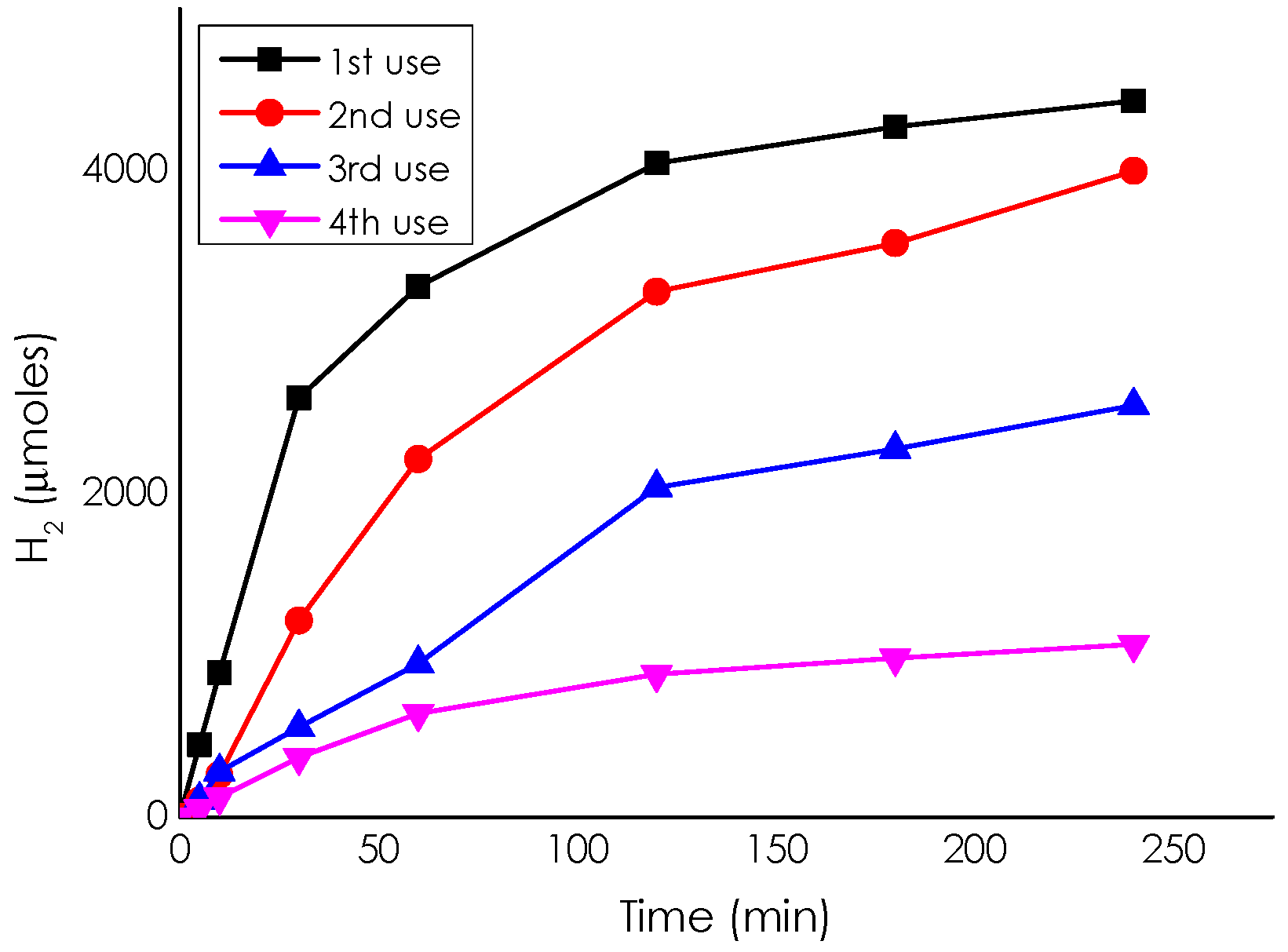
Catalytic Activity
3. Materials and Methods
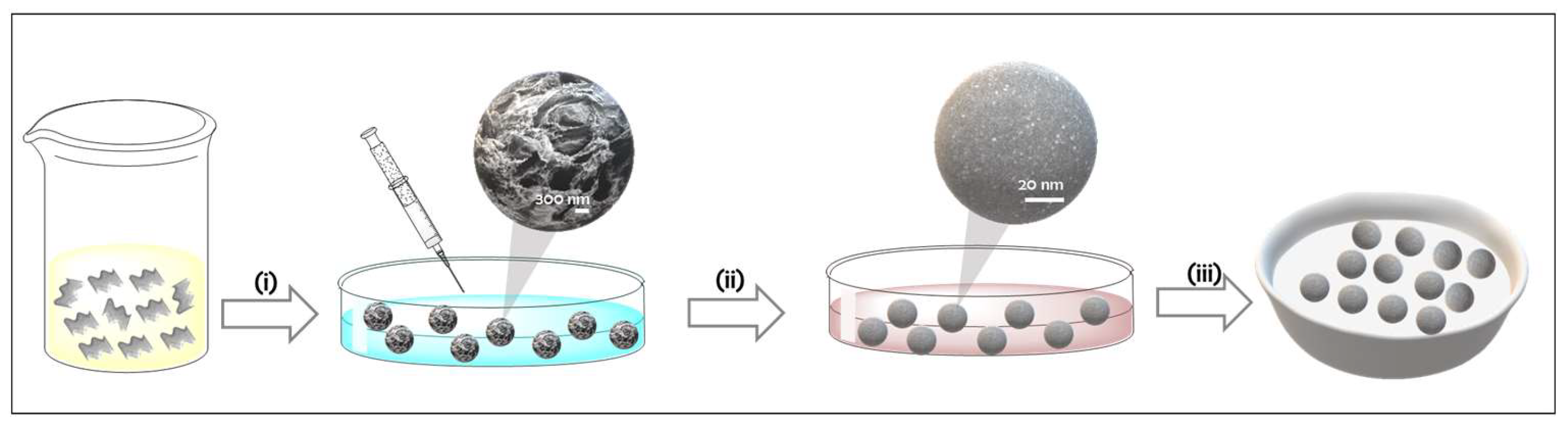
3.1. Synthesis of Pd/CS Aerogels and Pd/CSGO Aerogels
3.2. Catalytic Activity Test
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Staffell, I.; Scamman, D.; Abad, A.V.; Balcombe, P.; Dodds, P.E.; Ekins, P.; Shah, N.; Ward, K.R. The role of hydrogen and fuel cells in the global energy system. Energy Environ. Sci. 2019, 12, 463–491. [Google Scholar] [CrossRef]
- Modisha, P.M.; Ouma, C.N.M.; Garidzirai, R.; Wasserscheid, P.; Bessarabov, D. The Prospect of Hydrogen Storage Using Liquid Organic Hydrogen Carriers. Energy Fuels 2019, 33, 2778–2796. [Google Scholar] [CrossRef]
- Sotoodeh, F.; Smith, K.J. An overview of the kinetics and catalysis of hydrogen storage on organic liquids. Can. J. Chem. Eng. 2013, 91, 1477–1490. [Google Scholar] [CrossRef]
- Zhong, H.; Iguchi, M.; Chatterjee, M.; Himeda, Y.; Xu, Q.; Kawanami, H. Formic Acid-Based Liquid Organic Hydrogen Carrier System with Heterogeneous Catalysts. Adv. Sustain. Syst. 2018, 2, 1700161. [Google Scholar] [CrossRef]
- Li, S.J.; Zhou, Y.T.; Kang, X.; Liu, D.X.; Gu, L.; Zhang, Q.H.; Yan, J.M.; Jiang, Q. A Simple and Effective Principle for a Rational Design of Heterogeneous Catalysts for Dehydrogenation of Formic Acid. Adv. Mater. 2019, 31, 1806781. [Google Scholar] [CrossRef]
- Boddien, A.; Mellmann, D.; Gärtner, F.; Jackstell, R.; Junge, H.; Dyson, P.J.; Laurenczy, G.; Ludwig, R.; Beller, M. Efficient Dehydrogenation of Formic Acid Using an Iron Catalyst, Science (Washington, DC, USA). Science 2011, 333, 1733–1736. [Google Scholar] [CrossRef] [PubMed]
- Akbayrak, S.; Tonbul, Y.; Özkar, S. Nanoceria supported palladium(0) nanoparticles: Superb catalyst in dehydrogenation of formic acid at room temperature. Appl. Catal. B Environ. 2017, 206, 384–392. [Google Scholar] [CrossRef]
- Bi, Q.-Y.; Lin, J.-D.; Liu, Y.-M.; He, H.-Y.; Huang, F.-Q.; Cao, Y. Dehydrogenation of Formic Acid at Room Temperature: Boosting Palladium Nanoparticle Efficiency by Coupling with Pyridinic-Nitrogen-Doped Carbon. Angew. Chem. 2016, 128, 12028–12032. [Google Scholar] [CrossRef]
- Li, Z.; Yang, X.; Tsumori, N.; Liu, Z.; Himeda, Y.; Autrey, T.; Xu, Q. Tandem Nitrogen Functionalization of Porous Carbon: Toward Immobilizing Highly Active Palladium Nanoclusters for Dehydrogenation of Formic Acid. ACS Catal. 2017, 7, 2720–2724. [Google Scholar] [CrossRef]
- Kandile, N.G.; Zaky, H.T.; Mohamed, M.I.; Nasr, A.S.; Ali, Y.G. Extraction and Characterization of Chitosan from Shrimp Shells. Open J. Org. Polym. Mater. 2018, 8, 33. [Google Scholar] [CrossRef]
- Molnár, Á. The use of chitosan-based metal catalysts in organic transformations. Coord. Chem. Rev. 2019, 388, 126–171. [Google Scholar] [CrossRef]
- Guibal, E. Heterogeneous catalysis on chitosan-based materials: A review. Prog. Polym. Sci. 2005, 30, 71–109. [Google Scholar] [CrossRef]
- El Kadib, A.; Primo, A.; Molvinger, K.; Bousmina, M.; Brunel, D. Nanosized Vanadium, Tungsten and Molybdenum Oxide Clusters Grown in Porous Chitosan Microspheres as Promising Hybrid Materials for Selective Alcohol Oxidation. Chem.-A Eur. J. 2011, 17, 7940–7946. [Google Scholar] [CrossRef] [PubMed]
- Barskiy, D.A.; Kovtunov, K.V.; Primo, A.; Corma, A.; Kaptein, R.; Koptyug, I.V. Selective Hydrogenation of 1,3-Butadiene and 1-Butyne over a Rh/Chitosan Catalyst Investigated by using Parahydrogen-Induced Polarization. ChemCatChem 2012, 4, 2031–2035. [Google Scholar] [CrossRef]
- Frindy, S.; Lahcini, M.; Primo, A.; Bousmina, M.; Garcia, H.; El Kadib, A. Pd embedded in chitosan microspheres as tunable soft-materials for Sonogashira cross-coupling in water–ethanol mixture. Green Chem. 2015, 17, 1893–1898. [Google Scholar] [CrossRef]
- Primo, A.; Quignard, F. Chitosan as efficient porous support for dispersion of highly active gold nanoparticles: Design of hybrid catalyst for carbon–carbon bond formation. Chem. Commun. 2010, 46, 5593. [Google Scholar] [CrossRef] [PubMed]
- Chtchigrovsky, M.; Primo, A.; Gonzalez, P.; Molvinger, K.; Robitzer, M.; Quignard, F.; Taran, F. Functionalized Chitosan as a Green, Recyclable, Biopolymer-Supported Catalyst for the [3 + 2] Huisgen Cycloaddition. Angew. Chem. Int. Ed. 2009, 48, 5916–5920. [Google Scholar] [CrossRef] [PubMed]
- Primo, A.; Liebel, M.; Quignard, F. Palladium Coordination Biopolymer: A Versatile Access to Highly Porous Dispersed Catalyst for Suzuki Reaction. Chem. Mater. 2009, 21, 621–627. [Google Scholar] [CrossRef]
- El Kadib, A. Chitosan as a sustainable organocatalyst: A concise overview. ChemSusChem 2015, 8, 217–244. [Google Scholar] [CrossRef]
- Bratskaya, S.; Privar, Y.; Nesterov, D.; Modin, E.; Kodess, M.I.; Slobodyuk, A.; Marinin, D.V.; Pestov, A.V. Chitosan Gels and Cryogels Cross-Linked with Diglycidyl Ethers of Ethylene Glycol and Polyethylene Glycol in Acidic Media. Biomacromolecules 2019, 20, 1635–1643. [Google Scholar] [CrossRef]
- El Kadib, A.; Bousmina, M.; Brunel, D. Recent progress in chitosan bio-based soft nanomaterials. J. Nanosci. Nanotechnol. 2014, 14, 308–331. [Google Scholar] [CrossRef] [PubMed]
- Frindy, S.; Primo, A.; Ennajih, H.; Qaiss, A.E.K.; Bouhfid, R.; Lahcini, M.; Essassi, E.M.; Garcia, H.; El Kadib, A. Chitosan–graphene oxide films and CO2-dried porous aerogel microspheres: Interfacial interplay and stability. Carbohydr. Polym. 2017, 167, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Valentin, R.; Molvinger, K.; Brunel, D. Supercritical CO2 dried chitosan: An efficient intrinsic heterogeneous catalyst in fine chemistry. New J. Chem. 2003, 27, 1690–1692. [Google Scholar] [CrossRef]
- Huang, T.; Shao, Y.-W.; Zhang, Q.; Deng, Y.-F.; Liang, Z.-X.; Guo, F.-Z.; Li, P.-C.; Wang, Y. Chitosan-Cross-Linked Graphene Oxide/Carboxymethyl Cellulose Aerogel Globules with High Structure Stability in Liquid and Extremely High Adsorption Ability. ACS Sustain. Chem. Eng. 2019, 7, 8775–8788. [Google Scholar] [CrossRef]
- Kolanthai, E.; Sindu, P.A.; Khajuria, D.K.; Veerla, S.C.; Kuppuswamy, D.; Catalani, L.H.; Mahapatra, D.R. Graphene Oxide—A Tool for the Preparation of Chemically Crosslinking Free Alginate–Chitosan–Collagen Scaffolds for Bone Tissue Engineering. ACS Appl. Mater. Interfaces 2018, 10, 12441–12452. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolna, Z.; Cerveny, L. Ammonium formate decomposition using palladium catalyst. Res. Chem. Intermed. 2000, 26, 489–497. [Google Scholar] [CrossRef]
- Han, D.; Yan, L. Supramolecular Hydrogel of Chitosan in the Presence of Graphene Oxide Nanosheets as 2D Cross-Linkers. ACS Sustain. Chem. Eng. 2013, 2, 296–300. [Google Scholar] [CrossRef]
- Wang, J.; Tan, H.; Jiang, D.; Zhou, K. Enhancing H2 evolution by optimizing H adatom combination and desorption over Pd nanocatalyst. Nano Energy 2017, 33, 410–417. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds Pd/CS, Pd/CS-GO1, Pd/CS-GO2 and Pd/CS-GO3 are available from the authors. |















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Pd (%) | N (%) | GO (%) | Surface Area (m2/g) | Pore Volume (cm3/g) | Pd NP Size (nm) |
|---|---|---|---|---|---|---|
| Pd/CS | 2.50 | 6.24 | - | 220 | 1.06 | 1.7 ± 0.5 |
| Pd/CS-GO1 | 3.78 | 6.79 | 3 | 459 | 1.45 | 1.6 ± 0.9 |
| Pd/CS-GO2 | 3.44 | 6.50 | 7 | 435 | 1.51 | 1.8 ± 0.8 |
| Pd/CS-GO3 | 1.53 | 6.54 | 12 | 333 | 1.31 | 1.7 ± 0.5 |
| CS-GO | - | 6.75 | 3 | 294 | 1.25 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anouar, A.; Katir, N.; El Kadib, A.; Primo, A.; García, H. Palladium Supported on Porous Chitosan–Graphene Oxide Aerogels as Highly Efficient Catalysts for Hydrogen Generation from Formate. Molecules 2019, 24, 3290. https://doi.org/10.3390/molecules24183290
Anouar A, Katir N, El Kadib A, Primo A, García H. Palladium Supported on Porous Chitosan–Graphene Oxide Aerogels as Highly Efficient Catalysts for Hydrogen Generation from Formate. Molecules. 2019; 24(18):3290. https://doi.org/10.3390/molecules24183290
Chicago/Turabian StyleAnouar, Aicha, Nadia Katir, Abdelkrim El Kadib, Ana Primo, and Hermenegildo García. 2019. "Palladium Supported on Porous Chitosan–Graphene Oxide Aerogels as Highly Efficient Catalysts for Hydrogen Generation from Formate" Molecules 24, no. 18: 3290. https://doi.org/10.3390/molecules24183290