Emergence and Applications of Base Metals (Fe, Co, and Ni) in Hydroboration and Hydrosilylation



Abstract
1. Introduction
2. Hydroboration
3. Iron-Catalyzed Hydroboration of Carbonyls
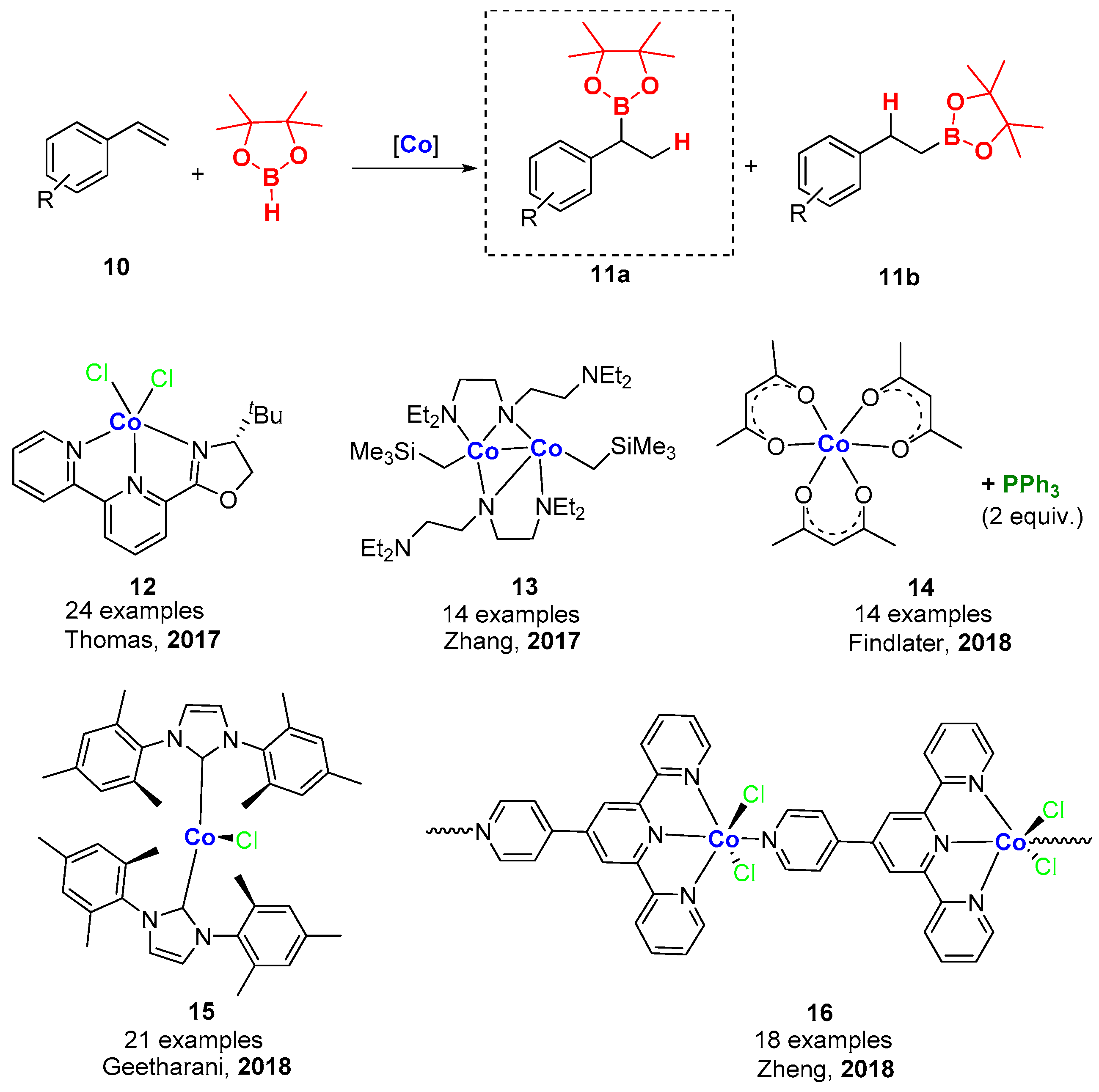
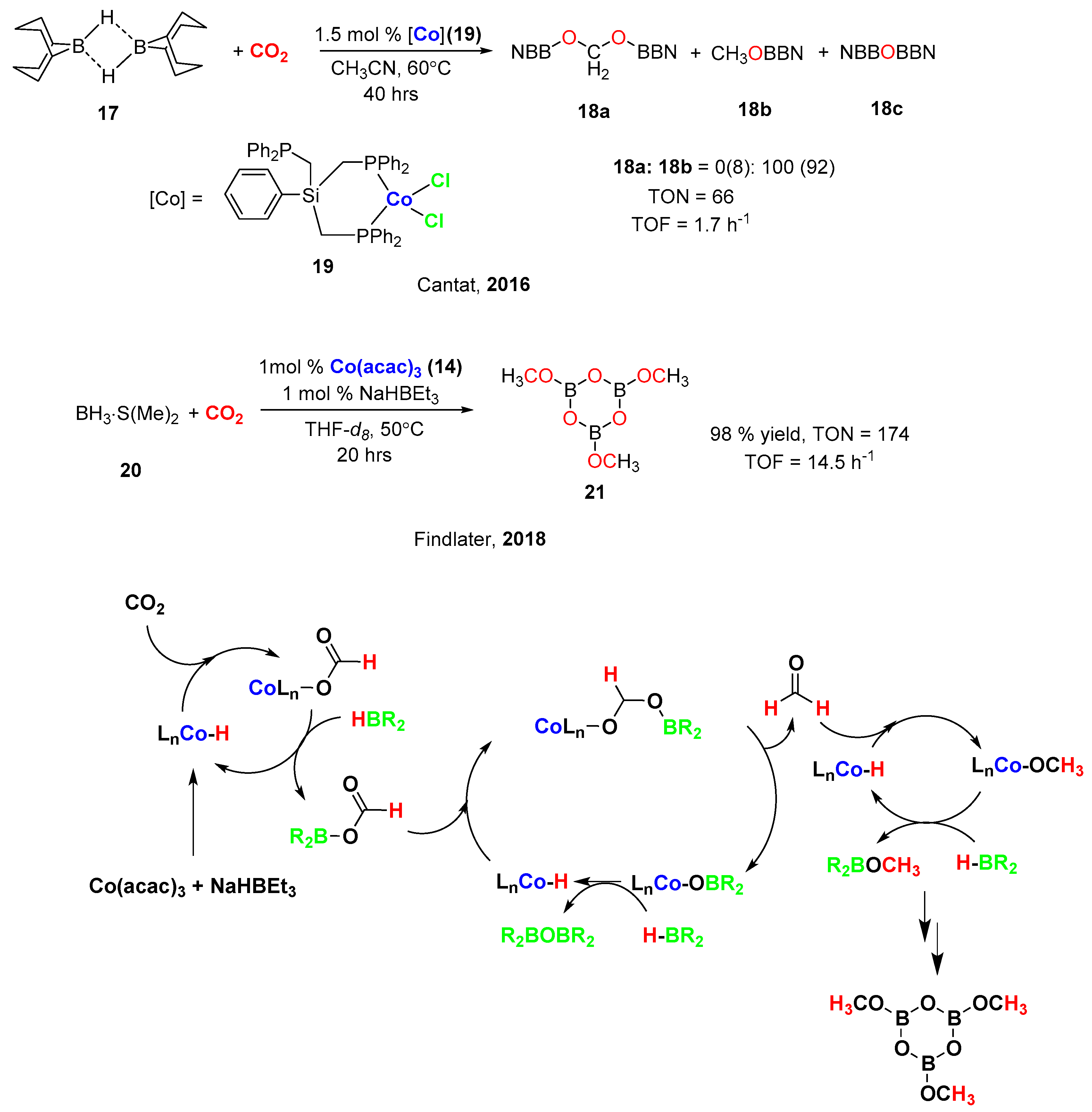
4. Cobalt-Catalyzed Hydroboration of Alkenes and CO2
5. Nickel-Catalyzed Hydroboration of N-Heteroarenes
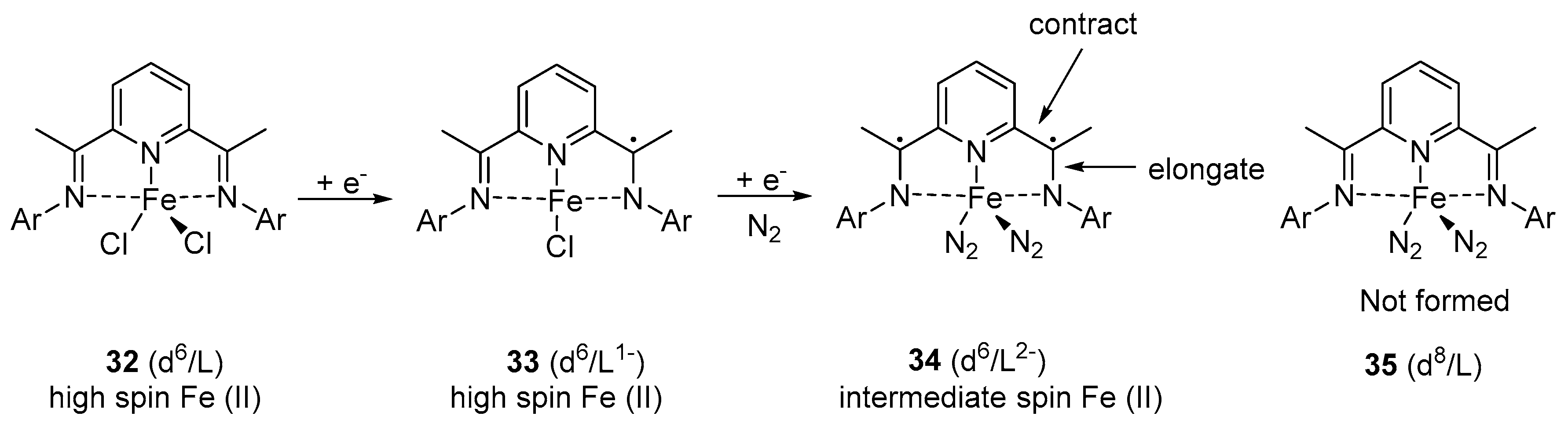
6. Iron-Catalyzed Hydrosilylation
7. Hydrosilylation Catalyzed by Iron- and Cobalt-Pincer Complexes
8. Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Magano, J.; Dunetz, J.R. Large-Scale Carbonyl Reductions in the Pharmaceutical Industry. Org. Process Res. Dev. 2012, 16, 1156–1184. [Google Scholar] [CrossRef]
- Zweig, J.E.; Kim, D.E.; Newhouse, T.R. Methods Utilizing First-Row Transition Metals in Natural Product Total Synthesis. Chem. Rev. 2017, 117, 11680–11752. [Google Scholar] [CrossRef] [PubMed]
- Hayler, J.D.; Leahy, D.K.; Simmons, E.M. A Pharmaceutical Industry Perspective on Sustainable Metal Catalysis. Organometallics 2019, 38, 36–46. [Google Scholar] [CrossRef]
- Seyferth, D.; Davison, A. The 1973 Nobel Prize for Chemistry. Science 1973, 182, 699–701. [Google Scholar] [CrossRef] [PubMed]
- Eliel, E.L.; Mosher, H.S. The 1975 Nobel Prize for Chemistry. Science 1975, 190, 772–774. [Google Scholar]
- Ault, A. The Nobel Prize in Chemistry for 2001. J. Chem. Educ. 2002, 79, 572–577. [Google Scholar] [CrossRef]
- Van Houten, J. A Century of Chemical Dynamics Traced through the Nobel Prizes. 1909: Wilhelm Ostwald. J. Chem. Educ. 2002, 79, 146–148. [Google Scholar] [CrossRef]
- Casey, C.P. 2005 Nobel Prize in Chemistry. Development of the Olefin Metathesis Method in Organic Synthesis. J. Chem. Educ. 2006, 83, 192–195. [Google Scholar] [CrossRef]
- Wisniak, J. The History of Catalysis: From the beginning to Nobel Prizes. Educ. Quim. 2010, 21, 60–69. [Google Scholar] [CrossRef]
- Wu, X.-F.; Anbarasan, P.; Neumann, H.; Beller, M. From Noble Metal to Nobel Prize: Palladium-Catalyzed Coupling Reactions as Key Methods in Organic Synthesis. Angew. Chem. Int. Ed. 2010, 49, 9047–9050. [Google Scholar] [CrossRef]
- Claverie, J.P.; Schaper, F. Ziegler-Natta Catalysis: 50 Years After the Nobel Prize. MRS Bull. 2013, 38, 213–218. [Google Scholar] [CrossRef]
- Chong, C.C.; Kinjo, R. Catalytic Hydroboration of Carbonyl Derivatives, Imines, and Carbon Dioxide. ACS Catal. 2015, 5, 3238–3259. [Google Scholar] [CrossRef]
- Díez-González, S.; Nolan, S.P. Transition Metal-Catalyzed Hydrosilylation of Carbonyl Compounds and Imines. A Review. Org. Prep. Proced. Int. 2007, 39, 523–559. [Google Scholar] [CrossRef]
- Riener, K.; Högerl, M.P.; Gigler, P.; Kühn, F.E. Rhodium-Catalyzed Hydrosilylation of Ketones: Catalyst Development and Mechanistic Insights. ACS Catal. 2012, 2, 613–621. [Google Scholar] [CrossRef]
- Alberico, D.; Scott, M.E.; Lautens, M. Aryl-Aryl Bond Formation by Transition Metal Catalyzed Direct Arylation. Chem. Rev. 2007, 107, 174–238. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L.; Vicente, R.; Kapdi, A.R. Transition-Metal-Catalyzed Direct Arylation of (Hetero)Arenes by C-H Bond Cleavage. Angew. Chem. Int. Ed. 2009, 48, 9792–9826. [Google Scholar] [CrossRef]
- Jiao, J.; Murakami, K.; Itami, K. Catlytic Methods for Aromatic C-H Amination: An Ideal Strategy for Nitrogen-based Functional Molecules. ACS Catal. 2016, 6, 610–633. [Google Scholar] [CrossRef]
- Park, Y.; Kim, Y.; Chang, S. Transition Metal-Catalyzed C-H Amination: Scope, Mechanism, and Applications. Chem. Rev. 2017, 117, 9247–9301. [Google Scholar] [CrossRef]
- Johnson, M. Available online: http://www.platinum.matthey.com/ (accessed on 30 August 2019).
- Daily Metal Prices. Available online: https://www.dailymetalprice.com/ (accessed on 30 August 2019).
- Albrecht, M.; Bedford, R.; Plietker, B. Catalytic and Organometallic Chemistry of Earth-Abundant Metals. Organometallics 2014, 33, 5619–5621. [Google Scholar] [CrossRef]
- White, M.C. Base Metal Catalysis: Embrace the Wild Side. Adv. Synth. Catal. 2016, 358, 2364–2365. [Google Scholar] [CrossRef]
- Campeau, L.-C.; Hazari, N. Cross-Coupling and Related Reactions: Connecting Past Success to the Development of New Reactions for the Future. Organometallics 2019, 38, 3–35. [Google Scholar] [CrossRef]
- Collet, F.; Dodd, R.H.; Dauban, P. Catalytic C-H Amination: Recent Progress and Future Directions. Chem. Commun. 2009, 5061–5074. [Google Scholar] [CrossRef] [PubMed]
- Collet, F.; Lescot, C.; Dauban, P. Catalytic C-H Amination: The stereoselectivity issue. Chem. Soc. Rev. 2011, 40, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Huang, Z. Advances in Base-Metal-Catalyzed Alkene Hydrosilylation. ACS Catalysis 2017, 7, 1227–1243. [Google Scholar] [CrossRef]
- Hofmann, R.J.; Vlatković, M.; Wiesbrock, F. Fifty Years of Hydrosilylation in Polymer Science: A Review of Current Trends of Low-Cost Transition-Metal and Metal-Free Catalysts, Non-Thermally Triggered Hydrosilylation Reactions, and Industrial Applications. Polymers 2017, 9, 534. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H. Borylation of Alkynes under Base/Coinage Metal Catalysis: Some Recent Developments. ACS Catal. 2016, 6, 1799–1811. [Google Scholar] [CrossRef]
- Chirik, P.J. Iron- and Cobalt-Catalyzed Alkene Hydrogenation: Catalysis with Both Redox-Active and Strong Field Ligands. Acc. Chem. Res. 2015, 48, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-Y.; Yu, S.-L.; Shen, W.-Y.; Gao, J.-X. Iron-, Cobalt-, and Nickel-Catalyzed Asymmetric Transfer Hydrogenation and Asymmetric Hydrogenation of Ketones. Acc. Chem. Res. 2015, 48, 2587–2598. [Google Scholar] [CrossRef] [PubMed]
- Alig, L.; Fritz, M.; Schneider, S. First Row Transition Metal (De)Hydrogenation Catalysis Based On Functional Pincer Ligands. Chem. Rev. 2019, 119, 2681–2751. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.S.; Liptrot, D.J.; Weetman, C. Alkaline Earths as main group reagents in molecular catalysis. Chem. Soc. Rev. 2016, 45, 972–988. [Google Scholar] [CrossRef]
- List, B. Introduction: Organocatalysis. Chem. Rev. 2007, 107, 5413–5415. [Google Scholar] [CrossRef]
- Alemán, J.; Cabrera, S. Applications of asymmetric organocatalysis in medicinal chemistry. Chem. Soc. Rev. 2013, 42, 774–793. [Google Scholar] [CrossRef] [PubMed]
- Díez-González, S.; Marion, N.; Nolan, S.P. N-Heterocyclic Carbenes in Late Transition Metal Catalysis. Chem. Rev. 2009, 109, 3612–3676. [Google Scholar] [CrossRef] [PubMed]
- Khusnutdinova, J.R.; Milstein, D. Metal-Ligand Cooperation. Angew. Chem. Int. Ed. 2015, 54, 12236–12273. [Google Scholar] [CrossRef] [PubMed]
- Junge, D.; Papa, V.; Beller, M. Cobalt-Pincer Complexes in Catalysis. Chem. Eur. J. 2019, 25, 122–143. [Google Scholar] [CrossRef] [PubMed]
- Bauer, G.; Hu, X. Recent developments of iron pincer complexes for catalytic applications. Inorg. Chem. Front. 2016, 3, 741–765. [Google Scholar] [CrossRef]
- Wei, D.; Darcel, C. Iron Catalysis in Reduction and Hydrometalation Reactions. Chem. Rev. 2019, 119, 2550–2610. [Google Scholar] [CrossRef]
- Obligacion, J.V.; Chirik, P.J. Earth-Abundant Transition Metal Catalysts for Alkene Hydrosilylation and Hydroboration: Opportunities and Assessments. Nat Rev Chem. 2018, 2, 15–34. [Google Scholar] [CrossRef]
- Suzuki, A. Cross-coupling reactions via organoboranes. J. Organomet. Chem. 2002, 653, 83–90. [Google Scholar] [CrossRef]
- Vitaliy, L.B.; Peter, S.S.; James, H.C.; Rafael, L. Industrial Applications of C-C Coupling Reactions. Curr. Org. Synth. 2010, 7, 614–627. [Google Scholar]
- Brown, H.C.; Rao, B.C.S. Hydroboration. II. A Remarkably Fast Addition of Diborane to Olefins-Scope and Stoichiometry of the Reaction. J. Am. Chem. Soc. 1959, 81, 6428–6434. [Google Scholar] [CrossRef]
- Evans, D.A.; Ratz, A.M.; Huff, B.E.; Sheppard, G.S. Total Synthesis of the Polyether Antibiotic Lonomycin A (Emericid). J. Am. Chem. Soc. 1995, 117, 3448–3467. [Google Scholar] [CrossRef]
- Yuan, K.; Suzuki, N.; Mellerup, S.K.; Wang, X.; Yamaguchi, S.; Wang, S. Pyridyl Directed Catalyst Free trans-Hydroboration of Internal Alkynes. Org. Lett. 2016, 18, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Matteson, D.S. Stereodirected Synthesis with Organoboranes, 1st ed.; Springer: Berlin/Heidelberg, Germany, 1995. [Google Scholar]
- Brands, K.M.J.; Kende, A.S. Remote substituent effects on regioselectivity in rhodium(I)-catalyzed hydroborations of norbornenes. Tetrahedron Lett. 1992, 33, 5887–5890. [Google Scholar] [CrossRef]
- Carroll, A.-M.; O’Sullivan, T.P.; Guiry, P.J. The Development of Enantioselective Rhodium-Catalyzed Hydroboration of Olefins. Adv. Synth. Catal. 2005, 347, 609–631. [Google Scholar] [CrossRef]
- Tamang, S.R.; Findlater, M. Iron Catalyzed Hydroboration of Aldehydes and Ketones. J. Org. Chem. 2017, 82, 12857–12862. [Google Scholar] [CrossRef] [PubMed]
- Tamang, S.R.; Bedi, D.; Shafiei-Haghighi, S.; Smith, C.R.; Crawford, C.; Findlater, M. Cobalt-Catalyzed Hydroboration of Alkenes, Aldehydes, and Ketones. Org. Lett. 2018, 20, 6695–6700. [Google Scholar] [CrossRef] [PubMed]
- Tamang, S.R.; Singh, A.; Unruh, D.K.; Findlater, M. Nickel-Catalyzed Regioselective 1,4-Hydroboration of N-Heteroarenes. ACS Catal. 2018, 8, 6186–6191. [Google Scholar] [CrossRef]
- Bai, T.; Janes, T.; Song, D. Homoleptic iron(II) and cobalt(II) bis(phosphoranimide) complexes for the selective hydrofunctionalization of unsaturated molecules. Dalton Trans. 2017, 46, 12408–12412. [Google Scholar] [CrossRef]
- Das, U.K.; Higman, C.S.; Gabidullin, B.; Hein, J.E.; Baker, R.T. Efficient and Selective Iron-Complex Catalyzed Hydroboration of Aldehydes. ACS Catal. 2018, 8, 1076–1081. [Google Scholar] [CrossRef]
- Blasius, C.K.; Vasilenko, V.; Gade, L.H. Ultrafast Iron-Catalyzed Reduction of Functionalized Ketones: Highly Enantioselective Synthesis of Halohydrines, Oxaheterocycles, and Aminoalcohols. Angew. Chem. Int. Ed. 2018, 57, 10231–10235. [Google Scholar] [CrossRef]
- Baishya, A.; Baruah, S.; Geetharani, K. Efficient Hydroboration of carbonyls by an iron(II) amide catalyst. Dalton Trans. 2018, 47, 9231–9236. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, E.; Cheng, J.; Zhang, G. Iron(II) coordination polymer catalyzed hydroboration of ketones. Dalton Trans. 2018, 47, 9579–9584. [Google Scholar] [CrossRef] [PubMed]
- Khoo, S.; Cao, J.; Ng, F.; So, C.-W. Synthesis of a Base-Stabilized Silicon(I)-Iron(II) Complex for Hydroboration of Carbonyl Compounds. Inorg. Chem. 2018, 57, 12452–12455. [Google Scholar] [CrossRef] [PubMed]
- Shegavi, M.L.; Baishya, A.; Geetharani, K.; Bose, S.K. Reusable Fe2O3-nanoparticle catalyzed efficient and selective hydroboration of carbonyl compounds. Org. Chem. Front. 2018, 5, 3520–3525. [Google Scholar] [CrossRef]
- MacNair, A.J.; Millet, C.R.P.; Nichol, G.S.; Ironmonger, A.; Thomas, S.P. Markovnikov-Selective, Activator-Free Iron-Catalyzed Vinylarene Hydroboration. ACS Catal. 2016, 6, 7217–7221. [Google Scholar] [CrossRef]
- Chen, X.; Cheng, Z.; Lu, Z. Iron-Catalyzed, Markovnikov-Selective Hydroboration of Styrenes. Org. Lett. 2017, 19, 969–971. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, M.D.; Thomas, S.P. Chemo-, regio-, and stereoselective iron-catalysed hydroboration of alkenes and alkynes. Chem. Commun. 2013, 49, 11230–11232. [Google Scholar] [CrossRef]
- Obligacion, J.V.; Chirik, P.J. Highly Selective Bis(imino)pyridine Iron-Catalyzed Alkene Hydroboration. Org. Lett. 2013, 15, 2680–2683. [Google Scholar] [CrossRef]
- Zhang, L.; Peng, D.; Leng, X.; Huang, Z. Iron-Catalyzed, Atom-Economical, Chemo- and Regioselective Alkene Hydroboration with Pinacolborane. Angew. Chem. Int. Ed. 2013, 52, 3676–3680. [Google Scholar] [CrossRef]
- Chen, J.; Xi, T.; Lu, Z. Iminopyridine Oxazoline Iron Catalyst for Asymmetric Hydroboration of 1,1-Disubstituted Aryl Alkenes. Org. Lett. 2014, 16, 6452–6455. [Google Scholar] [CrossRef]
- Zheng, J.; Sortais, J.-B.; Darcel, C. [(NHC)Fe(CO)4] Efficient Pre-catalyst for Selective Hydroboration of Alkenes. ChemCatChem 2014, 6, 763–766. [Google Scholar] [CrossRef]
- Tseng, K.-N.T.; Kampf, J.W.; Szymczak, N.K. Regulation of Iron-Catalyzed Olefin Hydroboration by Ligand Modifications at a Remote Site. ACS Catal. 2015, 5, 411–415. [Google Scholar] [CrossRef]
- Espinal-Viguri, M.; Woof, C.R.; Webster, R.L. Iron-Catalyzed Hydroboration: Unlocking Reactivity through Ligand Modulation. Chem. Eur. J. 2016, 22, 11605–11608. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Song, H.; Zhuang, X.; Tung, C.-H.; Wang, W. Iron-Catalyzed 1,2-elective Hydroboration of N-Heteroarenes. J. Am. Chem. Soc. 2017, 139, 17775–17778. [Google Scholar] [CrossRef] [PubMed]
- Haberberger, M.; Enthaler, S. Straightforward Iron-Catalyzed Synthesis of Vinylboronates by the Hydroboration of Alkynes. Chem. Asian. J. 2013, 8, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Gorgas, N.; Alves, L.G.; Stöger, B.; Martins, A.M.; Veiros, L.F.; Kirchner, K. Stable, Yet Highly Reactive Nonclassical Iron(II) Polyhydride Pincer Complexes: Z-Selective Dimerization and Hydroboration of Terminal Alkynes. J. Am. Chem. Soc. 2017, 139, 8130–8133. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Kato, T.; Nishibayashi, Y. Hydroboration of Alkynes Catalyzed by Pyrrolide-Based PNP Pincer-Iron Complexes. Org. Lett. 2017, 19, 4323–4326. [Google Scholar] [CrossRef]
- Cahiez, G.; Moyeux, A. Cobalt-Catalyzed Cross-Coupling Reactions. Chem. Rev. 2010, 110, 1435–1462. [Google Scholar] [CrossRef]
- Pellissier, H.; Clavier, H. Enantioselective Cobalt-Catalyzed Transformations. Chem. Rev. 2014, 114, 2775–2823. [Google Scholar] [CrossRef]
- Mukherjee, A.; Milstein, D. Homogeneous Catalysis by Cobalt and Manganese Pincer Complexes. ACS Catal. 2018, 8, 11435–11469. [Google Scholar] [CrossRef]
- Ai, W.; Zhong, R.; Liu, X.; Liu, Q. Hydride Transfer Reactions Catalyzed by Cobalt Complexes. Chem. Rev. 2019, 119, 2876–2953. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, D.; Entsminger, S.W.; Fout, A.R. Insights into a Chemoselective Cobalt Catalyst for the Hydroboration of Alkenes and Nitriles. ACS Catal. 2017, 7, 3730–3734. [Google Scholar] [CrossRef]
- Ben-Daat, H.; Rock, C.L.; Flores, M.; Groy, T.L.; Bowman, A.C.; Trovitch, R.J. Hydroboration of alkynes and nitriles using an a-diimine cobalt hydride catalyst. Chem. Commun. 2017, 53, 7333–7336. [Google Scholar] [CrossRef] [PubMed]
- Cruz, T.F.C.; Lopes, P.S.; Pereira, L.C.J.; Veiros, L.F.; Gomes, P.T. Hydroboration of Terminal Olefins with Pinacolborane Catalyzed by New Mono(2-Iminopyrrolyl) Cobalt(II) Complexes. Inorg. Chem. 2018, 57, 8146–8159. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Chen, J.; Lu, Z. Cobalt-catalyzed asymmetric hydroboration of aryl ketones with pinacolborane. Chem. Commun. 2015, 51, 5725–5727. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zeng, H.; Cheng, J.; Zheng, S.; Golen, J.A.; Manke, D.R.; Zhang, G. Cobalt(II) Coordination Polymer as a Precatalyst for Selective Hydroboration of Aldehydes, Ketones, and Imines. J. Org. Chem. 2018, 83, 9442–9448. [Google Scholar] [CrossRef] [PubMed]
- Obligacion, J.V.; Chirik, P.J. Bis(imino)pyridine Cobalt-Catalyzed Alkene Isomerizaion-Hydroboration: A Strategy for Remote Hydrofunctionalization with Terminal Selectivity. J. Am. Chem. Soc. 2013, 135, 19107–19110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zuo, Z.; Leng, X.; Huang, Z. A Cobalt-Catalyzed Alkene Hydroboration with Pinacolborane. Angew. Chem. Int. Ed. 2014, 53, 2696–2700. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zuo, Z.; Wan, X.; Huang, Z. Cobalt-Catalyzed Enantioselective Hydroboration of 1,1-Disubstituted Aryl Alkenes. J. Am. Chem. Soc. 2014, 136, 15501–15504. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wu, J.; Wang, M.; Zeng, H.; Cheng, J.; Neary, M.C.; Zheng, S. Cobalt-Catalyzed Regioselective Hydroboration of Terminal Alkenes. Eur. J. Org. Chem. 2017, 2017, 5814–5818. [Google Scholar] [CrossRef]
- Cruz, T.F.C.; Veiros, L.F.; Gomes, P.T. Cobalt(I) Complexes of 5-Aryl-2-iminopyrrolyl Ligands: Synthesis, Spin Isomerism, and Application in Catalytic Hydroboration. Inorg. Chem. 2018, 57, 14671–14685. [Google Scholar] [CrossRef] [PubMed]
- Macaulay, C.M.; Gustafson, S.J.; Fuller, J.T.; Kwon, D.-H.; Ogawa, T.; Ferguson, M.J.; McDonald, R.; Lumsden, M.D.; Bischof, S.M.; Sydora, O.L.; et al. Alkene Isomerization-Hydroboration Catalyzed by First-Row Transition-Metal (Mn, Fe, Co, and Ni) N-Phosphinoamidinate Complexes: Origin of Reactivity and Selectivity. ACS Catal. 2018, 8, 9907–9925. [Google Scholar] [CrossRef]
- Pang, M.; Wu, C.; Zhuang, X.; Zhang, F.; Su, M.; Tong, Q.; Tung, C.-H.; Wang, W. Addition of a B-H Bond across an Amido-Cobalt Bond: Co(II)-H-Catalyzed Hydroboration of Olefins. Organometallics 2018, 37, 1462–1467. [Google Scholar] [CrossRef]
- Palmer, W.N.; Diao, T.; Pappas, I.; Chirik, P.J. High-Activity Cobalt Catalysts for Alkene Hydroboration with Electronically Responsive Terpyridine and a-Diimine Ligands. ACS Catal. 2015, 5, 622–626. [Google Scholar] [CrossRef]
- Reilly, S.W.; Webster, C.E.; Hollis, T.K.; Valle, H.U. Transmetallation from CCC-NHC pincer Zr complexes in the synthesis of air-stable CCC-NHC pince Co(III) complexes and initial hydroboration trials. Dalton Trans. 2016, 45, 2823–2828. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Docherty, J.H.; Dominey, A.P.; Thomas, S.P. Cobalt-catalysed Markovnikov selective hydroboration of vinylarenes. Chem. Commun. 2017, 53, 4726–4729. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.K.; Sethulekshmi, A.S.; Geetharani, K. Markovnikov-Selective Co(I)-Catalyzed Hydroboration of Vinylarenes and Carbonyl Compounds. Org. Lett. 2018, 20, 7840–7845. [Google Scholar] [CrossRef]
- Zhang, G.; Wu, J.; Li, S.; Cass, S.; Zheng, S. Markovnikov-Selective Hydroboration of Vinylarenes Catlyzed by a Cobalt(II) Coordination Polymer. Org. Lett. 2018, 20, 7893–7897. [Google Scholar] [CrossRef]
- Docherty, J.H.; Peng, J.; Dominey, A.P.; Thomas, S.P. Activation and discovery of earth-abundant metal catalysts using sodium tert-butoxide. Nat. Chem. 2017, 9, 595–600. [Google Scholar] [CrossRef]
- Tamang, S.R.; Findlater, M. Cobalt catalyzed reduction of CO2 via hydroboration. Dalton Trans. 2018, 47, 8199–8203. [Google Scholar] [CrossRef]
- Aloisi, A.; Berthet, J.-C.; Genre, C.; Thuéry, P.; Cantat, T. Complexes of the tripodal phosphine ligands PhSi(XPPh2)3 (X = CH2, O): Synthesis, structure and catalytic activity in the hydroboration of CO2. Dalton Trans. 2016, 45, 14774–14788. [Google Scholar] [CrossRef] [PubMed]
- Tasker, S.Z.; Standley, E.A.; Jamison, T.F. Recent Advances in Homogeneous Nickel Catalysis. Nature 2014, 509, 299. [Google Scholar] [CrossRef] [PubMed]
- Ananikov, V.P. Nickel: The “Spirited Horse” of Transition Metal Catalysis. ACS Catal. 2015, 5, 1964–1971. [Google Scholar] [CrossRef]
- Chakraborty, S.; Zhang, J.; Krause, J.A.; Guan, H. An Efficient Nickel Catalyst for the Reduction of Carbon Dioxide with a Borane. J. Am. Chem. Soc. 2010, 132, 8872–8873. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Meng, W.; Ma, Q.-Q.; Zhang, J.; Li, H.; Li, S.; Zhao, Q.; Chen, X. Hydroboration of CO2 catalyzed by bis(phosphinite) pincer ligated nickel thiolate complexes. Dalton Trans. 2017, 46, 4504–4509. [Google Scholar] [CrossRef] [PubMed]
- Murphy, L.J.; Hollenhorst, H.; McDonald, R.; Ferguson, M.; Lumsden, M.D.; Turculet, L. Selective Ni-Catalyzed Hydroboration of CO2 to the Formaldehyde Level Enabled by New PSiP Ligation. Organometallics 2017, 36, 3709–3720. [Google Scholar] [CrossRef]
- Zhang, J.; Chang, J.; Liu, T.; Cao, B.; Ding, Y.; Chen, X. Application of POCOP Pincer Nickel Complexes to the Catalytic Hydroboration of Carbon Dioxide. Catalysts 2018, 8, 508. [Google Scholar] [CrossRef]
- Espinosa, M.R.; Charboneau, D.J.; Garcia de Oliveira, A.; Hazari, N. Controlling Selectivity in the Hydroboration of Carbon Dioxide to the Formic Acid, Formaldehyde, and Methanol Oxidation Levels. ACS Catal. 2019, 9, 301–314. [Google Scholar] [CrossRef]
- Chakraborty, S.; Bhattacharya, P.; Dai, H.; Guan, H. Nickel and Iron Pincer Complexes as Catalysts for the Reduction of Carbonyl Compounds. Acc. Chem. Res. 2015, 48, 1995–2003. [Google Scholar] [CrossRef]
- King, A.E.; Stieber, S.C.E.; Henson, N.J.; Kozimor, S.A.; Scott, B.L.; Smythe, N.C.; Sutton, A.D.; Gordon, J.C. Ni(bpy)(cod): A Convenient Entryway into the Efficient Hydroboration of Ketones, Aldehydes, and Imines. Eur. J. Inorg. Chem. 2016, 2016, 1635–1640. [Google Scholar] [CrossRef]
- Nakamura, G.; Nakajima, Y.; Matsumoto, K.; Srinivas, V.; Shimada, S. Nitrile Hydroboration reations catalyzed by simple nickel salts, bis(acetylacetonato)nickel(II) and its derivatives. Catal. Sci. Technol. 2017, 7, 3196–3199. [Google Scholar] [CrossRef]
- Kerchner, H.A.; Montgomery, J. Synthesis of Secondary and Tertiary Alklboranes via Formal Hydroboration of Terminal and 1,1-Disubstituted Alkenes. Org. Lett. 2016, 18, 5760–5763. [Google Scholar] [CrossRef] [PubMed]
- Touney, E.E.; Van Hoveln, R.; Buttke, C.T.; Freidberg, M.D.; Guzei, I.A.; Schomaker, J.M. Heteroleptic Nickel Complexes for the Markovnikov-Selective Hydroboration of Styrenes. Organometallics 2016, 35, 3436–3439. [Google Scholar] [CrossRef]
- Iwai, T.; Harada, T.; Shimada, H.; Asano, K.; Sawamura, M. A Polystyrene-Cross-Linking Bisphosphine: Controlled Metal Monochelation and Ligand-Enabled First-Row Transition Metal Catalysis. ACS Catal. 2017, 7, 1681–1692. [Google Scholar] [CrossRef]
- Li, J.-F.; Wei, Z.-Z.; Wang, Y.-Q.; Ye, M. Base-free nickel-catalyzed hydroboration of simple alkenes with bis(pinacolato)diboron in an alcoholic solvent. Green Chem. 2017, 19, 4498–4502. [Google Scholar] [CrossRef]
- Cozzi, P.; Carganico, G.; Fusar, D.; Grossoni, M.; Menichincheri, M.; Pinciroli, V.; Tonani, R.; Vaghi, F.; Salvati, P. Imidazol-1-yl and pyridin-3-yl derivatives of 4-phenyl-1,4-dihydropyridines combining Ca2+ antagonism and thromboxane A2 synthase inhibition. J. Med. Chem. 1993, 36, 2964–2972. [Google Scholar] [CrossRef] [PubMed]
- Lavilla, R. Recent developments in the chemistry of dihydropyridines. J. Chem. Soc. Perkin Trans. 2002, 1141–1156. [Google Scholar] [CrossRef]
- Arrowsmith, M.; Hill, M.S.; Hadlington, T.; Kociok-Köhn, G.; Weetman, C. Magnesium-Catalyzed Hydroboration of Pyridines. Organometallics 2011, 30, 5556–5559. [Google Scholar] [CrossRef]
- Kaithal, A.; Chatterjee, B.; Gunanathan, C. Ruthenium-Catalyzed Regioselective 1,4-Hydroboration of Pyridines. Org. Lett. 2016, 18, 3402–3405. [Google Scholar] [CrossRef]
- Speier, J.L.; Webster, J.A.; Barnes, G.H. The Addition of Silicon Hydrides to Olefinic Double Bonds. Part II. The Use of Group VII Metal Catalysts. J. Am. Chem. Soc. 1957, 79, 974–979. [Google Scholar] [CrossRef]
- Karstedt, B.D. General Electric Company. U.S. Patent US3775452A, 1973. [Google Scholar]
- Markó, E.; Stérin, S.; Buisine, O.; Mignani, G.; Branlard, P.; Tinant, B.; Declercq, J.-P. Selective and Efficient Platinum())-Carbene Complexes as Hydrosilylation Catalysts. Science 2002, 298, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.N.; Stein, J.; Gao, Y.; Colborn, R.E.; Hutchins, G. Platinum Catalysts Used in the Silicones Industry. Platinum Metals Rev. 1997, 41, 66. [Google Scholar]
- Troegel, D.; Stohrer, J. Recent advances and actual challenges in late transition metal catalyzed hydrosilylation of olefins from an industrial point of view. Coord. Chem. Rev. 2011, 255, 1440–1459. [Google Scholar] [CrossRef]
- Meister, T.K.; Riener, K.; Gigler, P.; Stohrer, J.; Herrmann, W.A.; Kühn, F.E. Platinum Catalysis Revisited-Unravleing Principles of Catalytic Olefin Hydrosilylation. ACS Catal. 2016, 6, 1274–1284. [Google Scholar] [CrossRef]
- Chen, J.; Guo, J.; Lu, Z. Recent Advances in Hydrometallation of Alkenes and Alkynes via the First Row Transition Metal Catalysis. Chin. J. Chem. 2018, 36, 1075–1109. [Google Scholar]
- Available online: https://www.springer.com/gp/book/9781402081712 (accessed on 26 August 2019).
- Wekesa, F.S.; Arias-Ugarte, R.; Kong, L.; Sumner, Z.; McGovern, G.P.; Findlater, M. Iron-Catalyzed Hydrosilylation of Aldehydes and Ketones under Solvent-Free Conditions. Organometallics 2015, 34, 5051–5056. [Google Scholar] [CrossRef]
- Tamang, S.R.; Cozzolino, A.F.; Findlater, M. Iron catalyzed selective reduction of esters to alcohols. Org. Biomol. Chem. 2019, 1834–1838. [Google Scholar] [CrossRef] [PubMed]
- Saini, A.; Smith, C.R.; Wekesa, F.S.; Helms, A.K.; Findlater, M. Conversion of aldimines to secondary amines using iron-catalysed hydrosilylation. Org. Biomol. Chem. 2018, 16, 9368–9372. [Google Scholar] [CrossRef] [PubMed]
- Butschke, B.; Fillman, K.L.; Bendikov, T.; Shimon, L.J.W.; Diskin-Posner, Y.; Leitus, G.; Gorelsky, S.I.; Neidig, M.L.; Milstein, D. How Innocent are Potentially Redox Non-Innocent Ligands? Electronic Structure and Metal Oxidation States in Iron-PNN Complexes as a Representative Case Study. Inorg. Chem. 2015, 54, 4909–4926. [Google Scholar] [CrossRef]
- Supej, M.J.; Volkov, A.; Darko, L.; West, R.A.; Darmon, J.M.; Schulz, C.E.; Wheeler, K.A.; Hoyt, H.M. Aryl-substituted BIAN complexes of iron dibromide: Synthesis, X-ray and electronic structure, and catalytic hydrosilylation activity. Polyhedron 2016, 114, 403–414. [Google Scholar] [CrossRef]
- Bart, S.C.; Chłopek, K.; Bill, E.; Bouwkamp, M.W.; Lobkovsky, E.; Neese, F.; Wieghardt, K.; Chirik, P.J. Electronic Structure of Bis(imino)pyridine Iron Dichloride, Monochloride, and Neutral Ligand Complexes: A Combined Structural, Spectroscopic, and Computational Study. J. Am. Chem. Soc. 2006, 128, 13901–13912. [Google Scholar] [CrossRef] [PubMed]
- Larson, P.J.; Wekesa, F.S.; Singh, A.; Smith, C.R.; Rajput, A.; McGovern, G.P.; Unruh, D.K.; Cozzolino, A.F.; Findlater, M. Synthesis, characterization, electrochemical properties and theoretical calculations of (BIAN) iron complexes. Polyhedron 2019, 159, 365–374. [Google Scholar] [CrossRef]
- Castro, L.C.M.; Sortais, J.-B.; Darcel, C. NHC-carbene cyclopentadienyl iron based catalyst for a general and efficient hydrosilylation of imines. Chem. Commun. 2012, 48, 151–153. [Google Scholar] [CrossRef]
- Bhunia, M.; Hota, P.K.; Vijaykumar, G.; Adhikari, D.; Mandal, S.K. A Highly Efficient Base-Metal Catalyst: Chemoselective Reduction of Imines to Amines Using An Abnormal-NHC-Fe(0) Complex. Organometallics 2016, 35, 2930–2937. [Google Scholar] [CrossRef]
- Li, B.; Zhang, S.; Wu, W.; Liang, L.; Jiang, S.; Chen, L.; Li, Y. Imidazolium-based ionic liquid-catalyzed hydrosilylation of imines and reductive amination of aldehydes using hydrosilane as the reductant. RSC Adv. 2017, 7, 31795–31799. [Google Scholar] [CrossRef]
- Bézier, D.; Venkanna, G.T.; Castro, L.C.M.; Zheng, J.; Roisnel, T.; Sortais, J.-B.; Darcel, C. Iron-Catalyzed hydrosilylation of Esters. Adv. Synth. Catal. 2012, 354, 1879–1884. [Google Scholar] [CrossRef]
- Das, S.; Li, Y.; Junge, K.; Beller, M. Synthesis of ethers from esters via Fe-catalyzed hydrosilylation. Chem. Commun. 2012, 48, 10742–10744. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Gregg, B.T.; Cutler, A.R. Catalytic Hydrosilylation of Organic Esters Using Manganese Carbonyl Acetate Complexes. J. Am. Chem. Soc. 1995, 117, 10139–10140. [Google Scholar] [CrossRef]
- Hansen, M.C.; Verdaguer, X.; Buchwald, S.L. Convenient Two-Step Conversion of Lactones into Cyclic Ethers. J. Org. Chem. 1998, 63, 2360–2361. [Google Scholar] [CrossRef]
- Matsubara, K.; Iura, T.; Maki, T.; Nagashima, H. A Triruthenium Carbonyl Cluster Bearing a Bridging Acenaphthylene Ligand: An Efficient Catalyst for Reduction of Esters, Carboxylic Acids, and Amides by Trialkylsilanes. J. Org. Chem. 2002, 67, 4985–4988. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Moriya, T.; Konakahara, T. An Efficient One-Pot Synthesis of Unsymmetrical Ethers: A Directly Reductive Deoxygenation of Esters Using an InBr3/Et3SiH Catalytic System. J. Org. Chem. 2007, 72, 5920–5922. [Google Scholar] [CrossRef] [PubMed]
- Junge, K.; Wendt, B.; Zhou, S.; Beller, M. Iron-Catalyzed Reduction of Carboxylic Esters to Alcohols. Eur. J. Org. Chem. 2013, 2013, 2061–2065. [Google Scholar] [CrossRef]
- Ruddy, J.; Kelly, C.M.; Crawford, S.M.; Wheaton, C.A.; Sydora, O.L.; Small, B.L.; Stradiotto, M.; Turculet, L. (N-Phosphinoamidinate)Iron Pre-Catalysts for the Room Temperature Hydrosilylation of Carbonyl Compounds with Broad Substrate Scope at Low Loadings. Organometallics 2013, 32, 5581–5588. [Google Scholar] [CrossRef]
- Farrar-Tobar, R.A.; Wozniak, B.; Savini, A.; Hinze, S.; Tin, S.; de Vries, J.G. Base-Free Iron Catalyzed Transfer Hydrogenation of Esters Using EtOH as Hydrogen Source. Angew. Chem. Int. Ed. 2019, 58, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Dai, H.; Bhattacharya, P.; Fairweather, N.T.; Gibson, M.S.; Krause, J.A.; Guan, H. Iron-Based Catalysts for the Hydrogenation of Esters to Alcohols. J. Am. Chem. Soc. 2014, 136, 7869–7872. [Google Scholar] [CrossRef] [PubMed]
- Dupau, P.; Tran Do, M.-L.; Gaillard, S.; Renaud, J.-L. Iron-Catalyzed Hydrogenation of Esters to Alcohols. Angew. Chem. Int. Ed. 2014, 53, 13004–13006. [Google Scholar] [CrossRef]
- Werkmeister, S.; Junge, K.; Wendt, B.; Alberico, E.; Jiao, H.; Baumann, W.; Junge, H.; Gallou, F.; Beller, M. Hydrogenation of Esters to Alcohols with a Well-Defined Iron Complex. Angew. Chem. Int. Ed. 2014, 53, 8722–8726. [Google Scholar] [CrossRef]
- Zell, T.; Ben-David, Y.; Milstein, D. Unprecedented Iron-Catalyzed Ester Hydrogenation. Mild, Selective, and Efficient Hydrogenation of Trifluoroacetic Esters to Alcohols Catalyzed by an Iron Pincer Complex. Angew. Chem. Int. Ed. 2014, 53, 4685–4689. [Google Scholar] [CrossRef]
- Zell, T.; Milstein, D. Hydrogenation and Dehydrogenation Iron Pincer Catalysts Capable of Metal-Ligand Cooperation by Aromatization/Dearomatization. Acc. Chem. Res. 2015, 48, 1979–1994. [Google Scholar] [CrossRef]
- Elangovan, S.; Wendt, B.; Topf, C.; Bachmann, S.; Scalone, M.; Spannenberg, A.; Jiao, H.; Baumann, W.; Junge, K.; Beller, M. Improved Second generation Iron Pincer Complexes for Effective Ester Hydrogenation. Adv. Synth. Catal. 2016, 358, 820–825. [Google Scholar] [CrossRef]
- Villa, M.; Miesel, D.; Hildebrandt, A.; Ragaini, F.; Schaarschmidt, D.; Jacobi von Wangelin, A. Synthesis and Catalysis of Redox-Active Bis(imino)acenaphthene (BIAN) Iron Complexes. ChemCatChem 2017, 9, 3203–3209. [Google Scholar] [CrossRef]
- Evans, D.A.; Cowley, A.H. Unique Radical Dearomatization and Two-Electron Reduction of a Redox-Active Ligand. J. Am. Chem. Soc. 2012, 134, 15672–15675. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Vargas-Baca, I.; Cowley, A.H. Sterically Directed Functionalization of the Redox-Active Bis(imino)acenaphthene Ligand Class: An Experimental and Theoretical Investigation. J. Am. Chem. Soc. 2013, 135, 13939–13946. [Google Scholar] [CrossRef]
- Fedushkin, I.L.; Skatova, A.A.; Chudakova, V.A.; Fukin, G.K. Four-Step Reduction of dpp-bian with Sodium Metal: Crystal Structures of the Sodium Salts of the Mono-, Di-, Tri- and Tetraanions of dpp-bian. Angew. Chem. Int. Ed. 2003, 42, 3294–3298. [Google Scholar] [CrossRef]
- Fedushkin, I.L.; Hummert, M.; Schumann, H. Molecular Structures and NMR Studies of Lithium and Germanium(II) Complexes of a New Chelating Amido-Imino Ligand Obtained by Addition of nBuLi to 1,2-Bis(arylimino)acenaphthene. Eur. J. Inorg. Chem. 2006, 2006, 3266–3273. [Google Scholar] [CrossRef]
- Smith, A.D.; Saini, A.; Singer, L.M.; Phadke, N.; Findlater, M. Synthesis, characterization and reactivity of iron- and cobalt-pincer complexes. Polyhedron 2016, 114, 286–291. [Google Scholar] [CrossRef]
- Asay, M.; Morales-Morales, D. Non-symmetric pincer ligands: Complexes and applications in catalysis. Dalton Trans. 2015, 44, 17432–17447. [Google Scholar] [CrossRef]
- Andrew, R.E.; González-Sebastián, L.; Chaplin, A.P. NHC-based pincer ligands: Carbenes with a bite. Dalton Trans. 2016, 45, 1299–1305. [Google Scholar] [CrossRef]
- Ananthnag, G.S.; Shetti, V.S. Synthesis, structure and catalysis of organometallic porphyrin-pincer hybrids: A review. Dalton Trans. 2017, 46, 14062–14082. [Google Scholar] [CrossRef]
- Balakrishna, M.S. Unusual and rare pincer ligands: Synthesis, metalation, reactivity and catalytic studies. Polyhedron 2018, 143, 2–10. [Google Scholar] [CrossRef]
- Lawrence, M.A.W.; Green, K.-A.; Nelson, P.N.; Lorraine, S.C. Review: Pincer ligands-Tunable, versatile and applicable. Polyhedron 2018, 143, 11–27. [Google Scholar] [CrossRef]
- Maser, L.; Vondung, L.; Langer, R. The ABC in pincer chemistry – From amine- to borylene- and carbon-based pincer-ligands. Polyhedron 2018, 143, 28–42. [Google Scholar] [CrossRef]
- Peris, E.; Crabtree, R.H. Key factors in pincer ligand design. Chem. Soc. Rev. 2018, 47, 1959–1968. [Google Scholar] [CrossRef] [PubMed]
- Tondreau, M.; Lobkovsky, E.; Chirik, P.J. Bis(imino)pyridine Iron Complexes for Aldehyde and Ketone Hydrosilylation. Org. Lett. 2008, 10, 2789–2792. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, P.; Krause, J.A.; Guan, H. Iron Hydride Complexes Bearing Phosphinite-Based Pincer Ligands: Synthesis, Reactivity, and Catalytic Apllication in Hydrosilylation Reactions. Organometallics 2011, 30, 4720–4729. [Google Scholar] [CrossRef]
- Bhattacharya, P.; Krause, J.A.; Guan, H. Activation of Dihydrogen and Silanes by Cationic Iron Bis(phosphinite) Pincer Complexes. Organometallics 2014, 33, 6113–6121. [Google Scholar] [CrossRef]
- Wu, S.; Li, X.; Xiong, Z.; Xu, W.; Lu, Y.; Sun, H. Synthesis and Reactivity of Silyl Iron, Cobalt, and Nickel Complexes Bearing a [PSiP]-Pincer Ligand via Si-H Bond Actviation. Organometallics 2013, 32, 3227–3237. [Google Scholar] [CrossRef]
- Zhao, H.; Sun, H.; Li, X. Synthesis and Catalytic Property of iron Pincer Complexes Generated by Csp3-H Activation. Organometallics 2014, 33, 3535–3539. [Google Scholar] [CrossRef]
- Huang, S.; Zhao, H.; Li, X.; Wang, L.; Sun, H. Synthesis of [POCOP]-pincer iron and cobalt complexes via Csp3-H activation and catalytic application of iron hydride in hydrosilylation reactions. RSC Adv. 2015, 5, 15660–15667. [Google Scholar] [CrossRef]
- Lin, H.-J.; Lutz, S.; O’Kane, C.; Zeller, M.; Chen, C.-H.; Al Assil, T.; Lee, W.-T. Synthesis and characterization of an iron complex bearing a hemilabile NNN-pincer for catalytic hydrsilylation of organic carbonyl compounds. Dalton Trans. 2018, 47, 3243–3247. [Google Scholar] [CrossRef] [PubMed]
- Sauer, D.C.; Wadepohl, H.; Gade, L.H. Cobalt Alkyl Complexes of a New Family of Chiral 1,3-Bis(2-pyridylimino)isoindolates and Their Application in Asymmetric Hydrosilylation. Inorg. Chem. 2012, 51, 12948–12958. [Google Scholar] [CrossRef]
- Zhou, H.; Sun, H.; Zhang, S.; Li, X. Synthesis and Reactivity of a Hydrido CNC Pincer Cobalt(III) Complex and Its Application in Hydrosilylation of Aldehydes and Ketones. Organometallics 2015, 34, 1479–1486. [Google Scholar] [CrossRef]
- Chen, X.; Lu, Z. Iminophenyl Oxazolinylphenylamine for Enantioselective Cobalt-Catalyzed Hydrosilylation of Aryl Ketones. Org. Lett. 2016, 18, 4658–4661. [Google Scholar] [CrossRef] [PubMed]
- Ott, J.C.; Wadepohl, H.; Enders, M.; Gade, L.H. Taking Solution Proton NMR to Its Extreme: Prediction and Detection of a Hydride Resonance in an Intermediate-Spin Iron Complex. J. Am. Chem. Soc. 2018, 140, 17413–17417. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, S.H.; Neidig, M.L. A Physical-Inorganic Approach for the Elucidation of Active Iron Species and Mechanism in Iron-Catalysed Cross-Coupling. Isr. J. Chem. 2017, 57, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Kneebone, J.L.; Brennessel, W.W.; Neidig, M.L. Intermediates and Reactivity in Iron-Catalyzed Cross-Couplings of Alkynyl Grignards with Alkyl Halides. J. Am. Chem. Soc. 2017, 139, 6988–7003. [Google Scholar] [CrossRef]
- Carpenter, S.H.; Baker, T.M.; Muñoz, S.B.; Brennessel, W.W.; Neidig, M.L. Multinuclear iron-phenyl species in reactions of simple iron salts with PhMgBr: Identification of Fe4(mu-Ph)6(THF)4 as a key reactive species for cross-coupling catalysis. Chem. Sci. 2018, 9, 7931–7939. [Google Scholar] [CrossRef]
- Sears, J.D.; Neate, P.G.N.; Neidig, M.L. Intermediates and Mechanism in Iron-Catalyzed Cross-Coupling. J. Am. Chem. Soc. 2018, 140, 11872–11883. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamang, S.R.; Findlater, M. Emergence and Applications of Base Metals (Fe, Co, and Ni) in Hydroboration and Hydrosilylation. Molecules 2019, 24, 3194. https://doi.org/10.3390/molecules24173194
Tamang SR, Findlater M. Emergence and Applications of Base Metals (Fe, Co, and Ni) in Hydroboration and Hydrosilylation. Molecules. 2019; 24(17):3194. https://doi.org/10.3390/molecules24173194
Chicago/Turabian StyleTamang, Sem Raj, and Michael Findlater. 2019. "Emergence and Applications of Base Metals (Fe, Co, and Ni) in Hydroboration and Hydrosilylation" Molecules 24, no. 17: 3194. https://doi.org/10.3390/molecules24173194
APA StyleTamang, S. R., & Findlater, M. (2019). Emergence and Applications of Base Metals (Fe, Co, and Ni) in Hydroboration and Hydrosilylation. Molecules, 24(17), 3194. https://doi.org/10.3390/molecules24173194