Prediction of Structure and Molecular Interaction with DNA of BvrR, a Virulence-Associated Regulatory Protein of Brucella

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
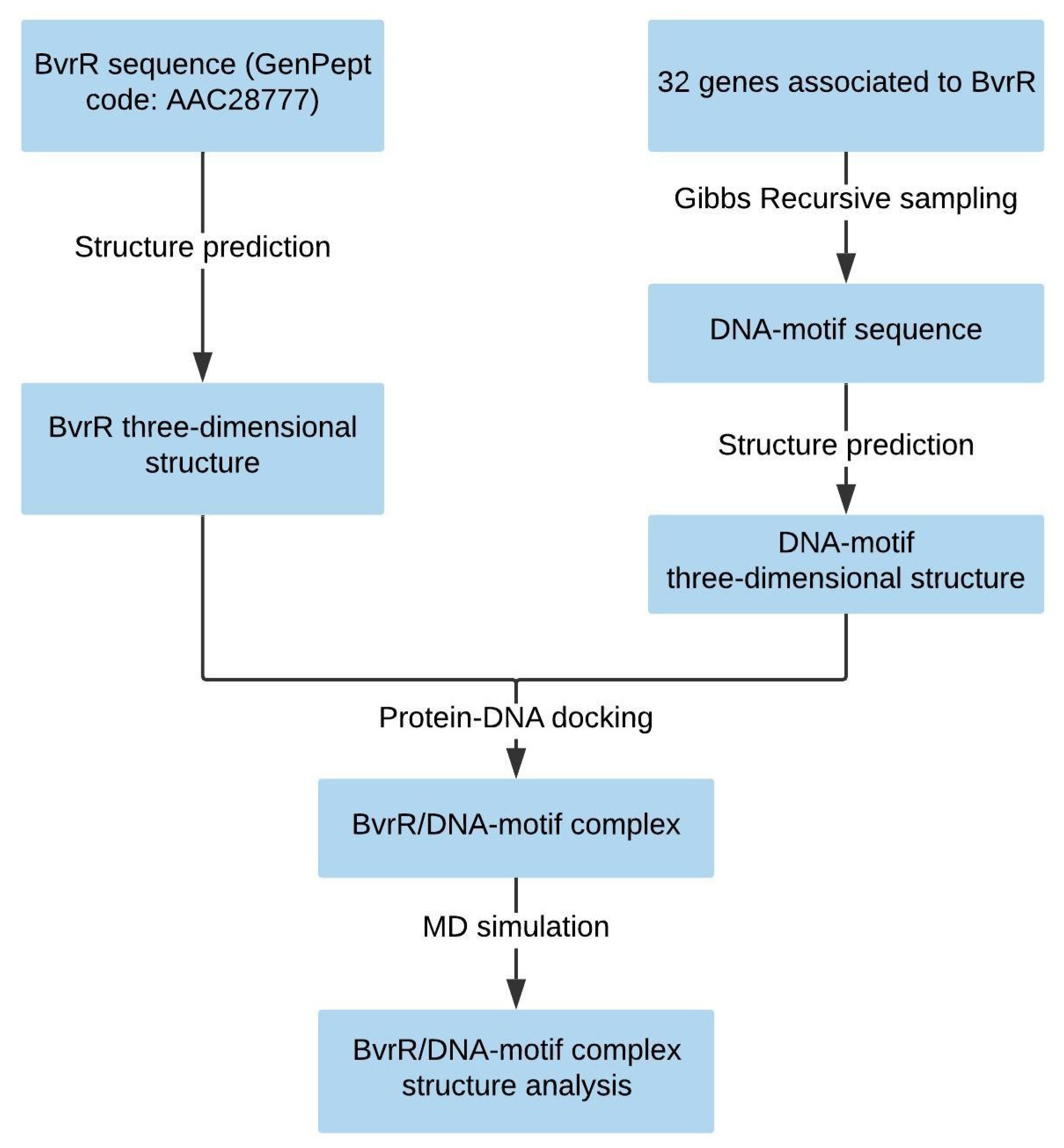
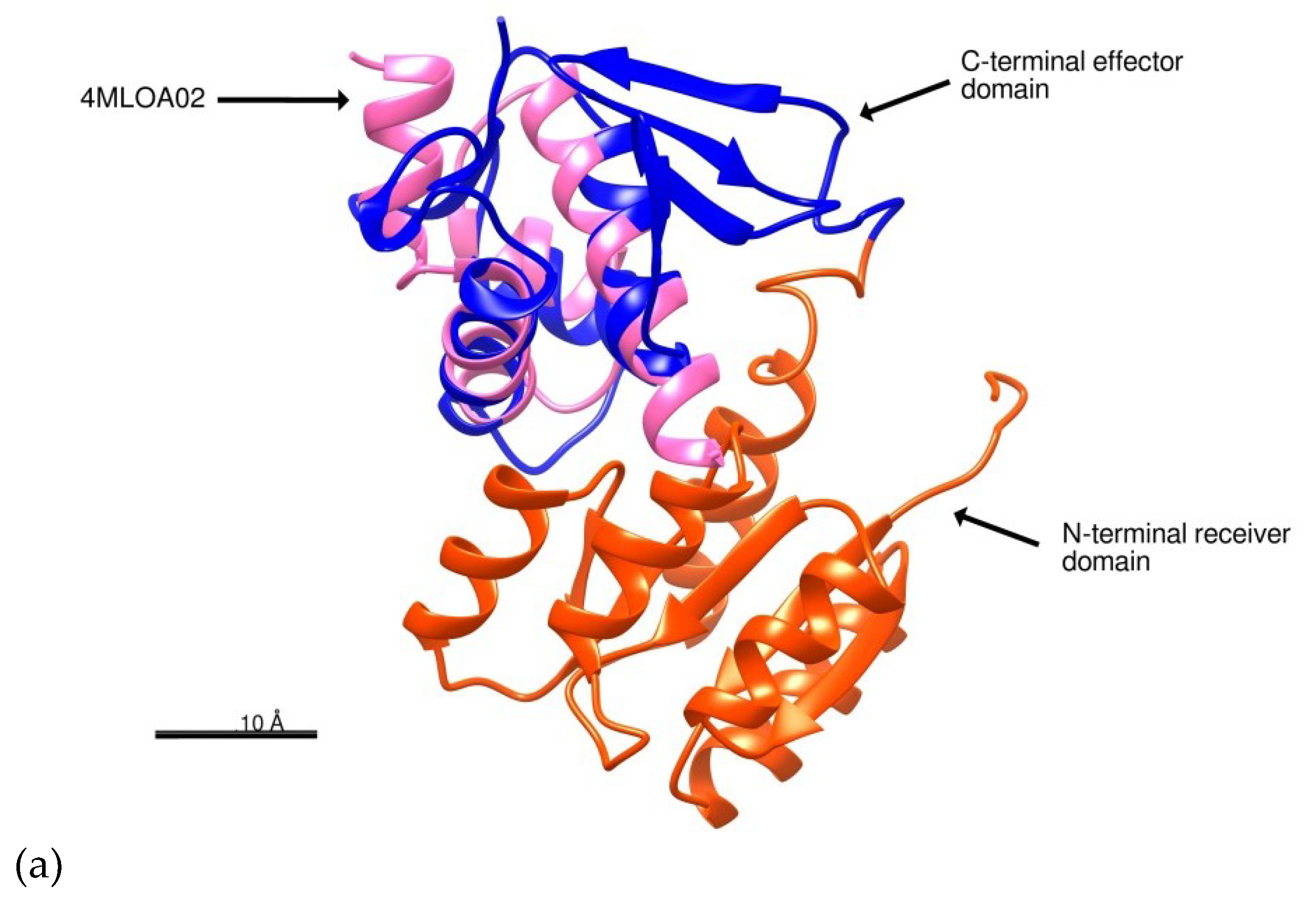
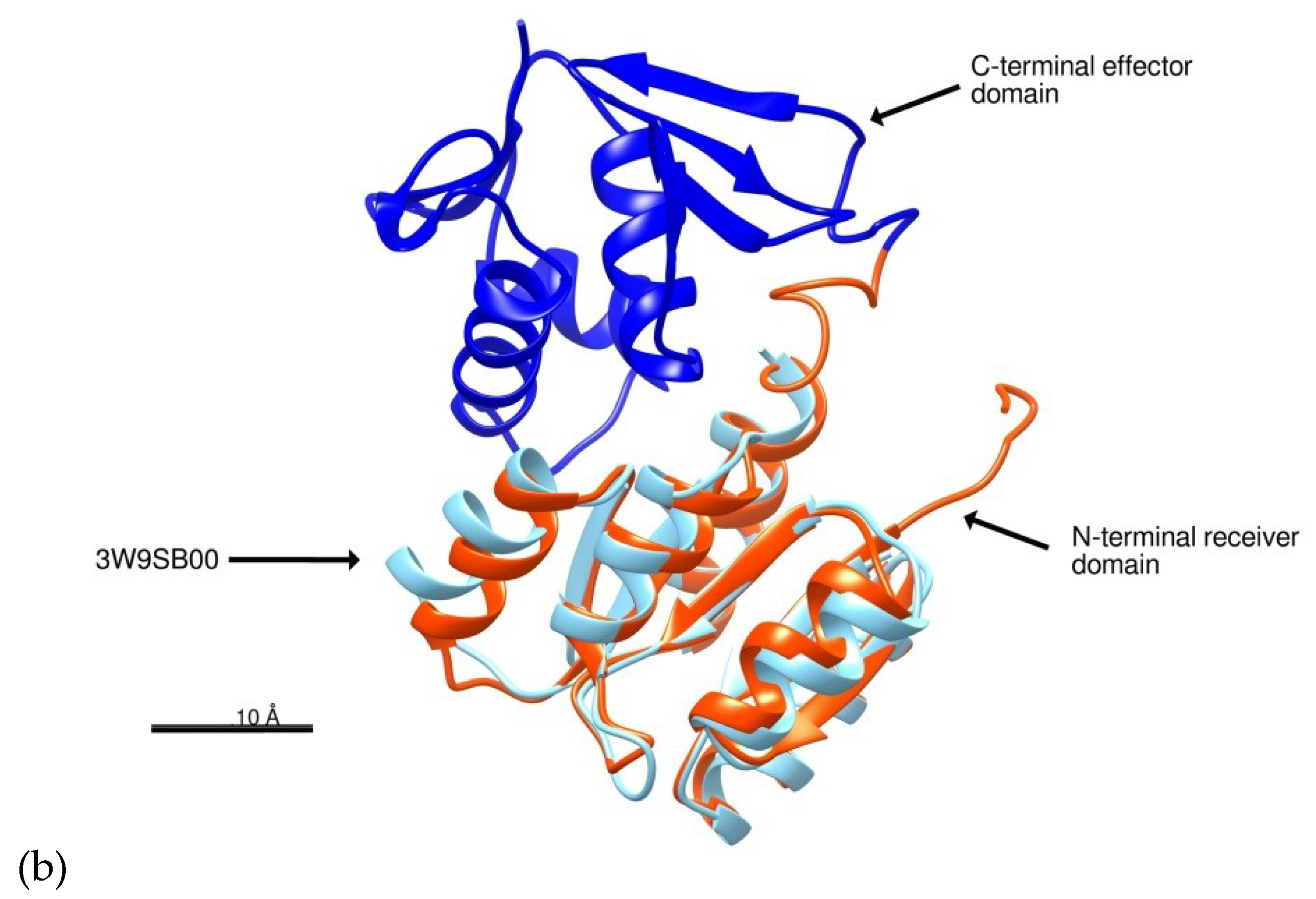
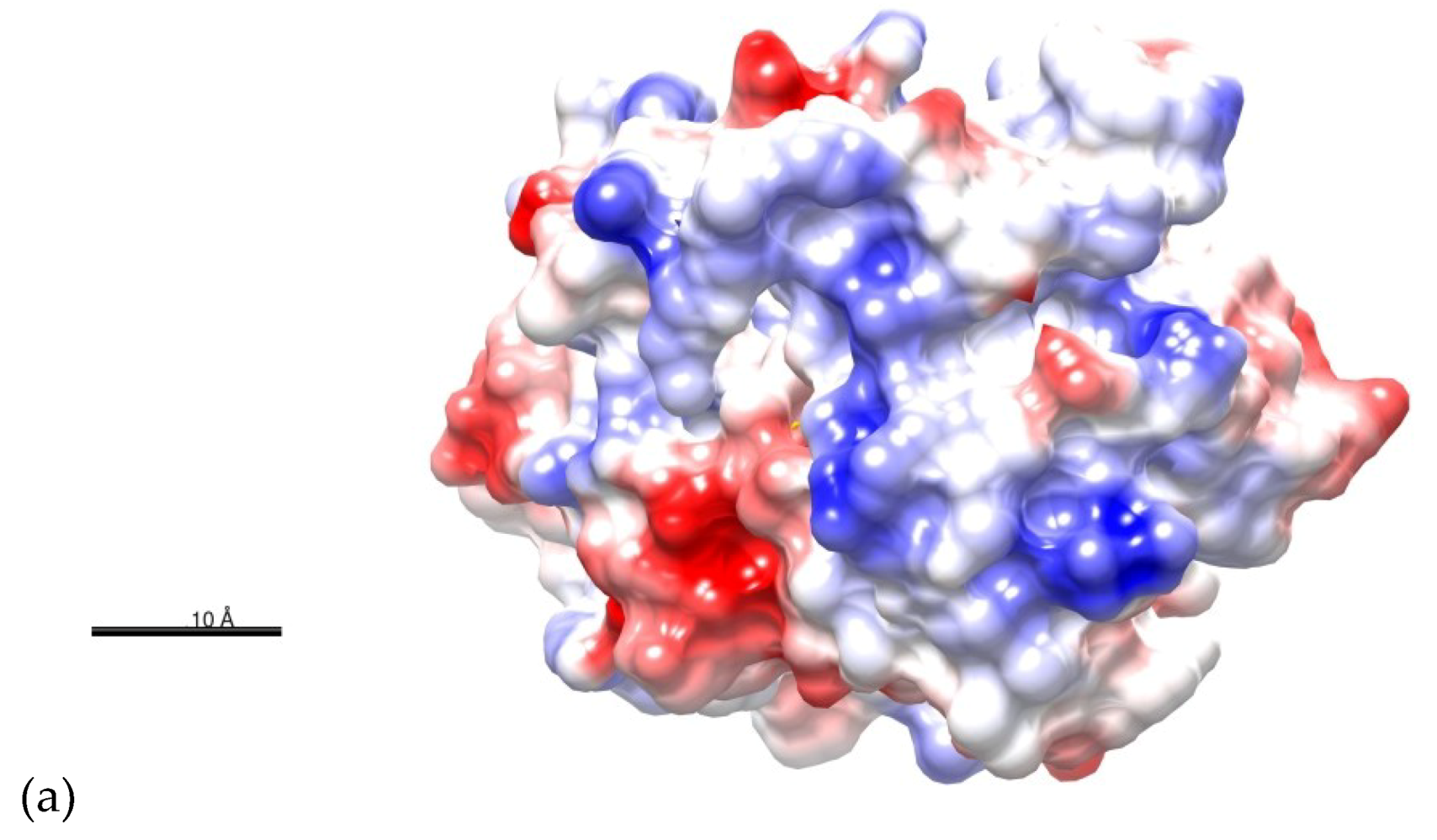
2.1. Structure Prediction of BvrR
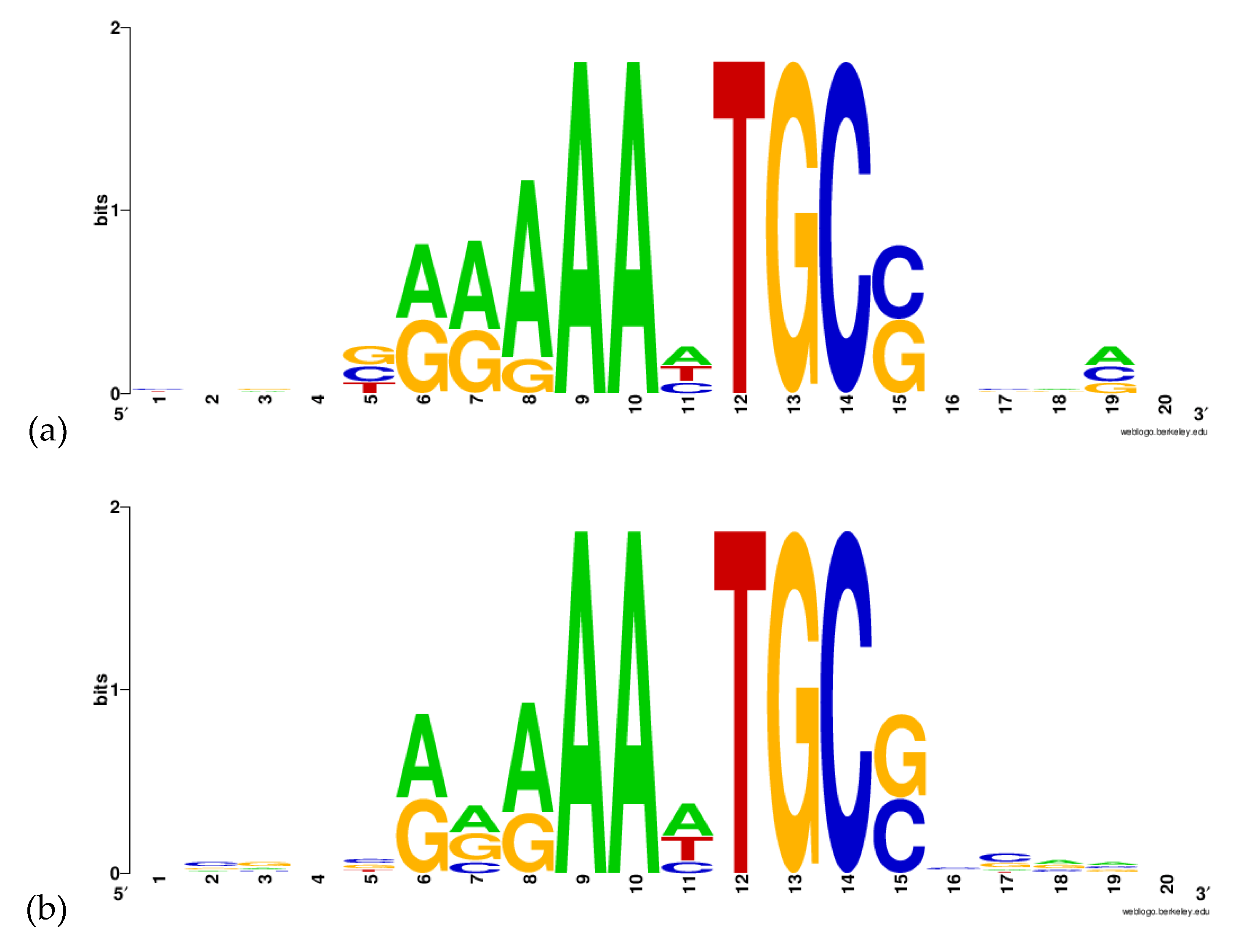
2.2. Prediction of DNA-Motif Recognized by BvrR
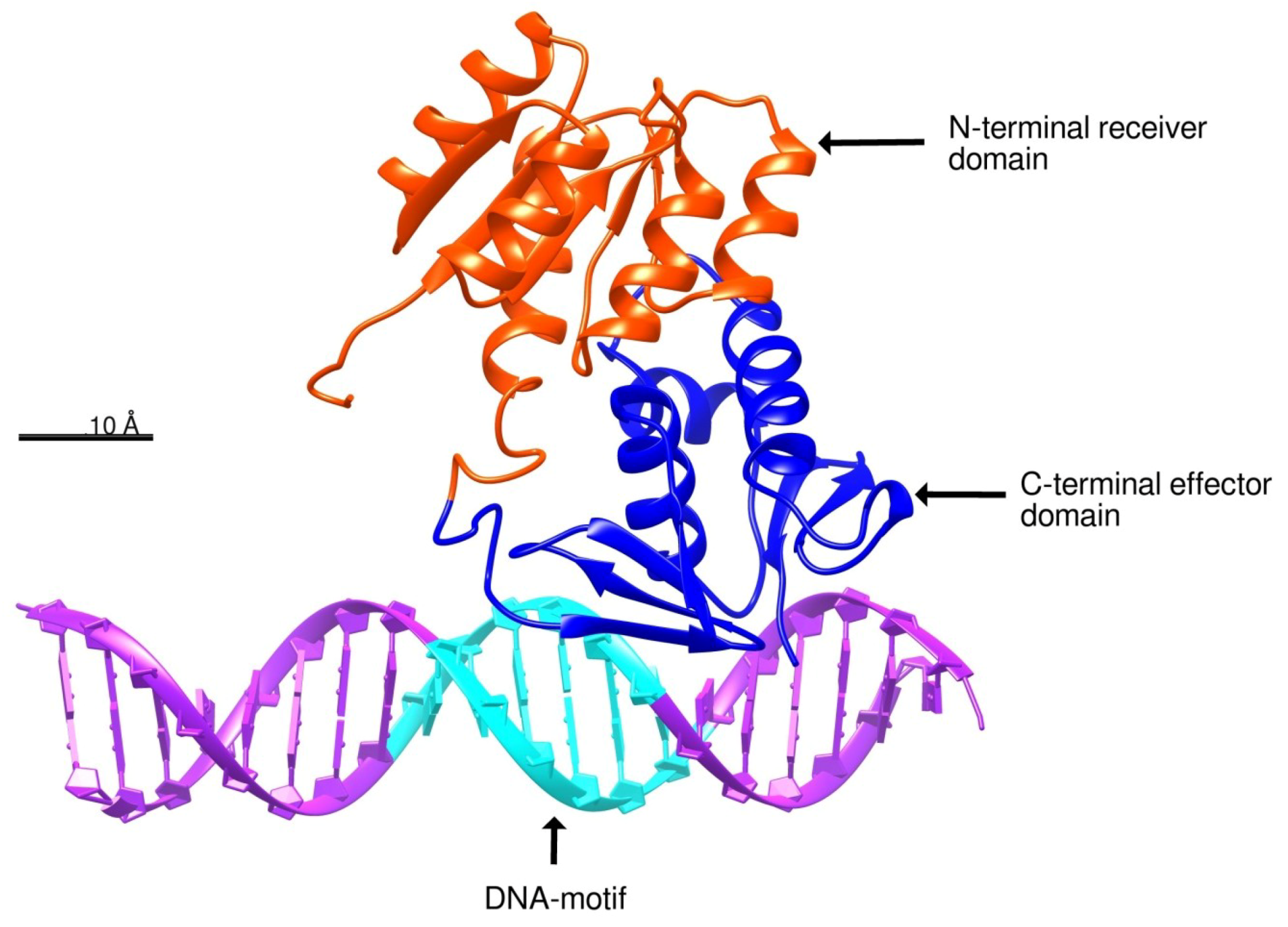
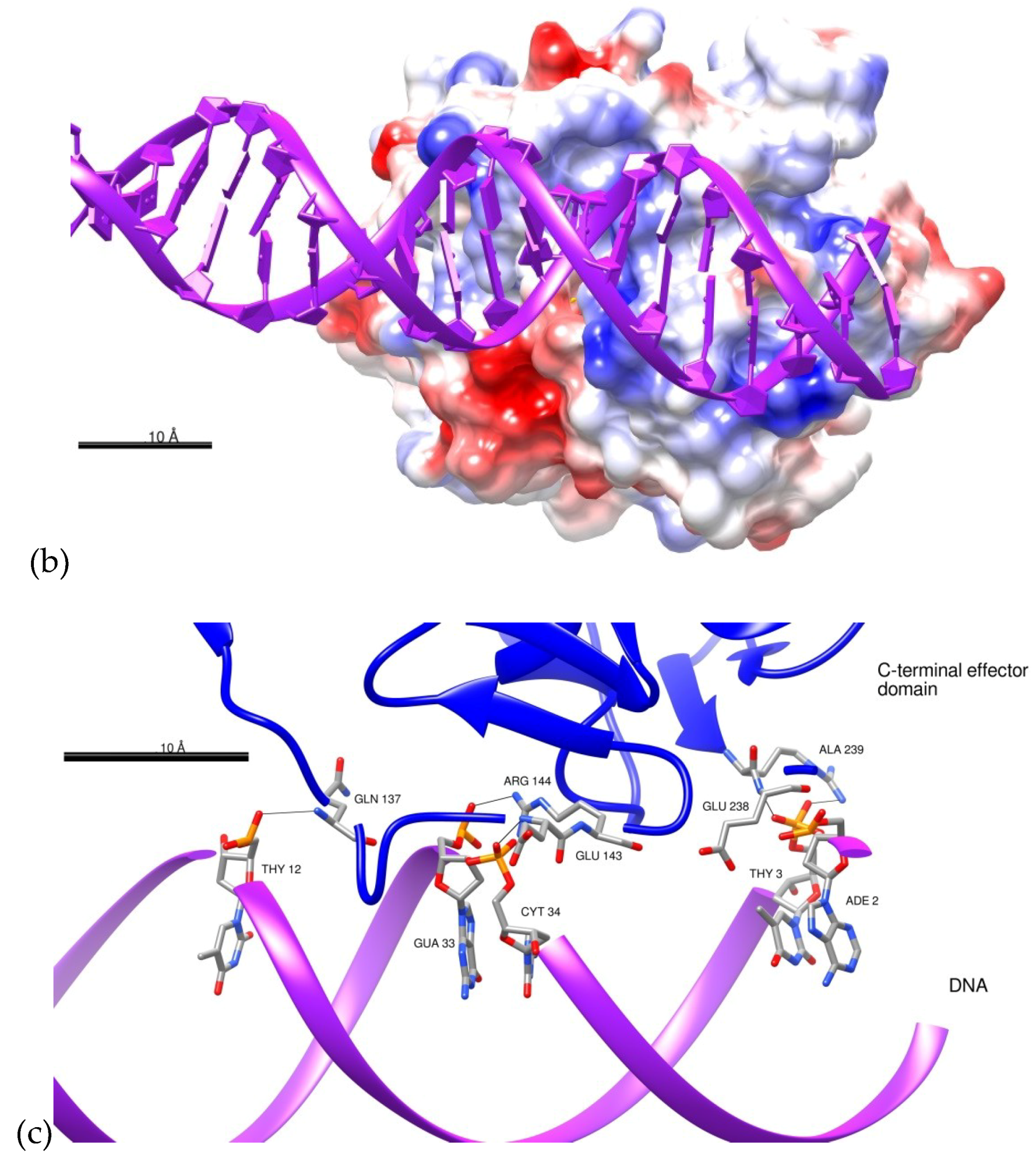
2.3. Docking of BvrR into the DNA-motif
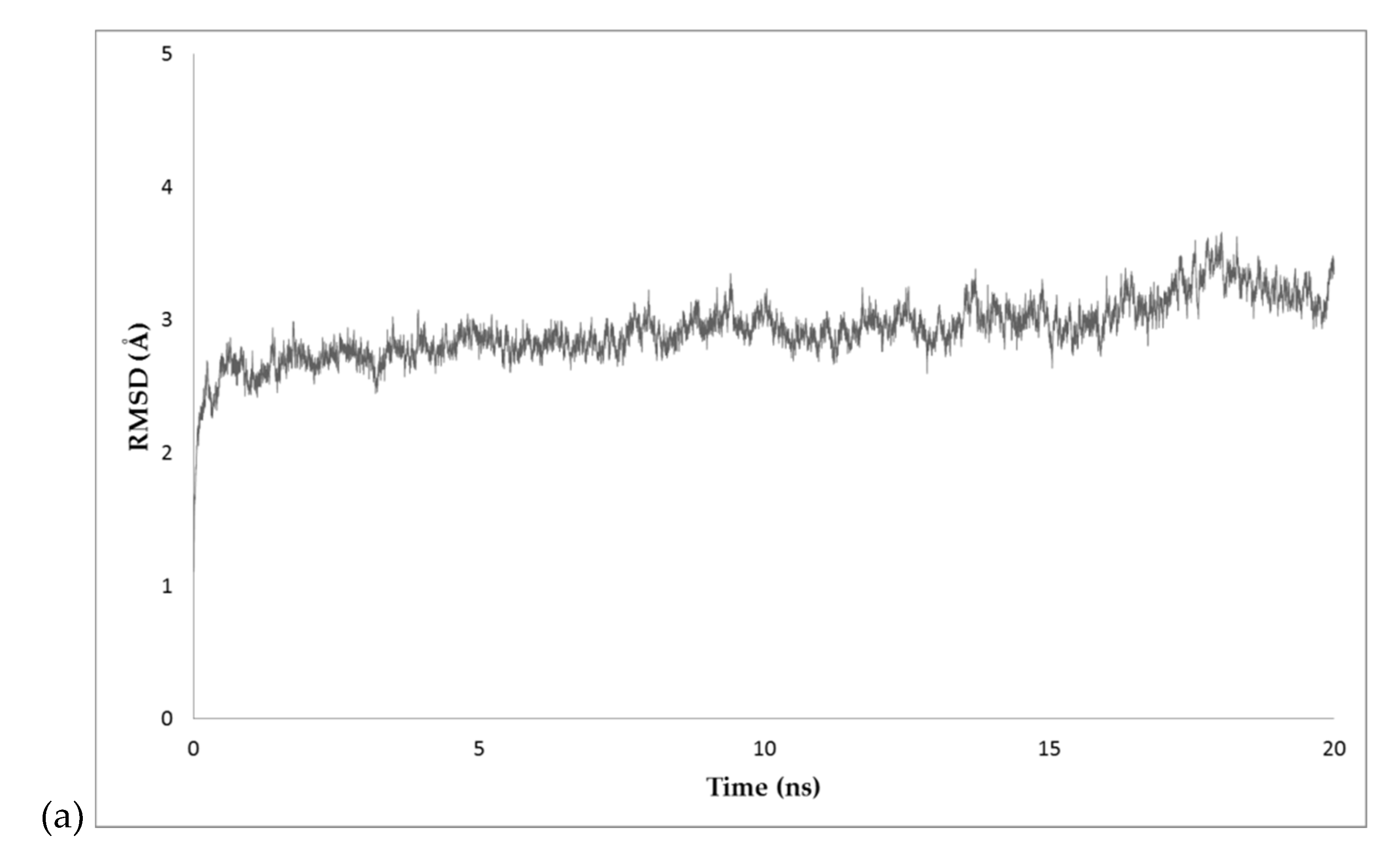
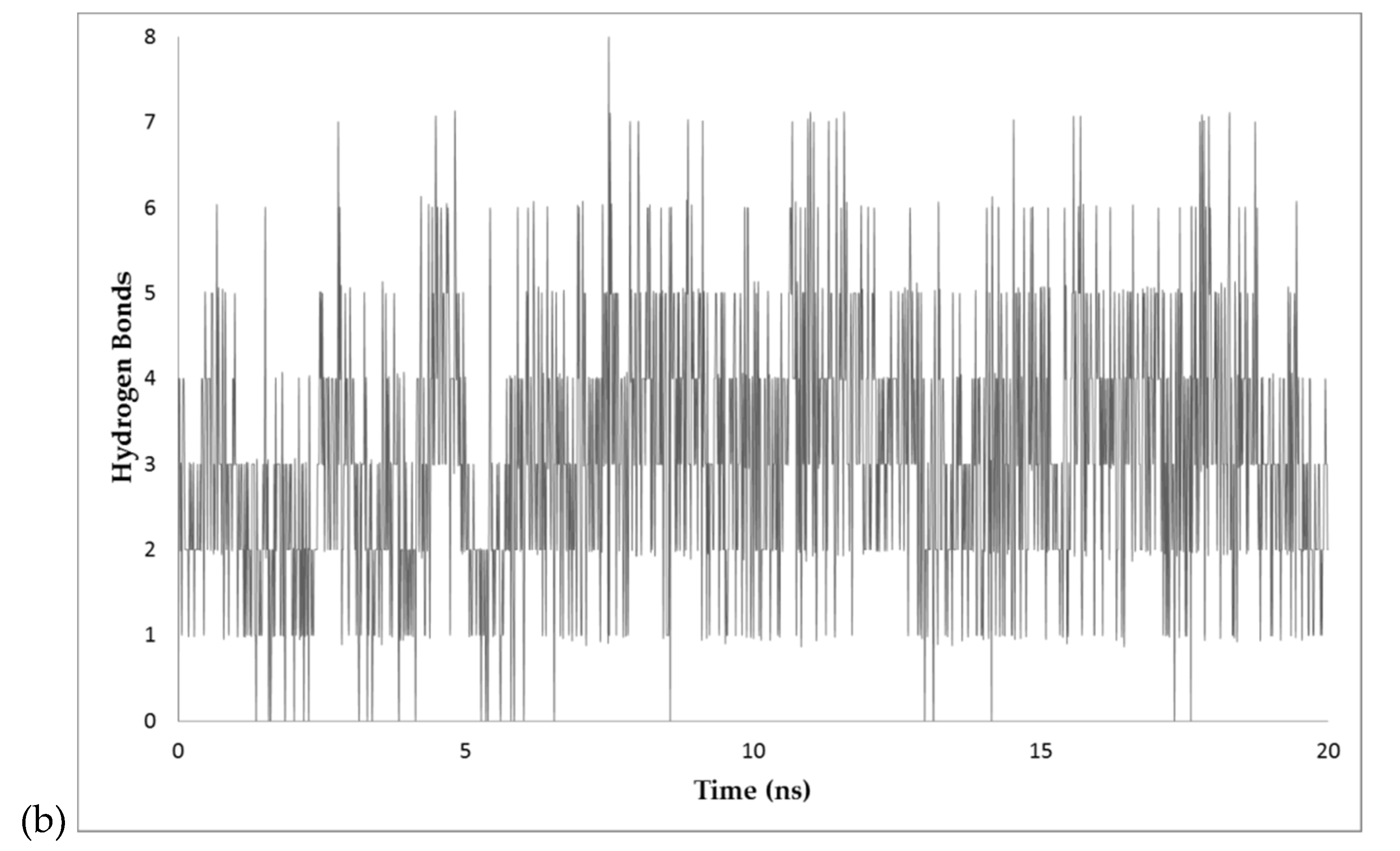
2.4. MD Simulations
3. Materials and Methods
3.1. Structure Prediction
3.2. Gibbs Sampling
3.3. Protein-DNA Docking
3.4. Molecular Dynamics Simulations
3.5. Analysis of Dynamics Simulations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ariza, J.; Bosilkovski, M.; Cascio, A.; Colmenero, J.D.; Corbel, M.J.; Falagas, M.E.; Memish, Z.A.; Roushan, M.R.H.; Rubinstein, E.; Sipsas, N.V.; et al. Perspectives for the Treatment of Brucellosis in the 21st Century: The Ioannina Recommendations. PLoS Med. 2007, 4, e317. [Google Scholar] [CrossRef] [PubMed]
- Corbel, M.J. Brucellosis in Humans and Animals; WHO Press: Geneva, Switzerland, 2006; pp. 1–102. [Google Scholar]
- Xavier, M.N.; Paixao, T.A.; den Hartigh, A.B.; Tsolis, R.M.; Santos, R.L. Pathogenesis of Brucella spp. Open Vet. Sci. J. 2010, 4, 109–118. [Google Scholar] [CrossRef]
- Bossi, P.; Tegnell, A.; Baka, A.; Van Loock, F.; Hendriks, J.; Werner, A.; Maidhof, H.; Gouvras, G. Task Force on Biological and Chemical Agent Threats, Public Health Directorate, European Commission, Luxembourg Bichat guidelines for the clinical management of brucellosis and bioterrorism-related brucellosis. Euro Surveill. 2004, 9, E15–E16. [Google Scholar] [CrossRef] [PubMed]
- Pappas, G.; Akritidis, N.; Bosilkovski, M.; Tsianos, E. Brucellosis. N. Engl. J. Med. 2005, 352, 2325–2336. [Google Scholar] [CrossRef] [PubMed]
- Fugier, E.; Pappas, G.; Gorvel, J.P. Virulence factors in brucellosis: Implications for aetiopathogenesis and treatment. Expert Rev. Mol. Med. 2007, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Acha, P.N.; Szyfres, B. Zoonosis y enfermedades transmisibles comunes al hombre y a los animales. vol. 1—Bacteriosis y micosis. Rev. Inst. Med. Trop. Sao Paulo 2001, 43, 338. [Google Scholar] [CrossRef]
- Murray, P.R.; Rosenthal, K.S.; Pfaller, M.A. Microbiologia Médica; Elsevier: Amsterdam, The Netherland, 2017; ISBN 9788491130765. [Google Scholar]
- Gorvel, J.P.; Moreno, E. Brucella intracellular life: From invasion to intracellular replication. Vet. Microbiol. 2002, 90, 281–297. [Google Scholar] [CrossRef]
- Watarai, M.; Kim, S.; Erdenebaatar, J.; Makino, S.; Horiuchi, M.; Shirahata, T.; Sakaguchi, S.; Katamine, S. Cellular prion protein promotes Brucella infection into macrophages. J. Exp. Med. 2003, 198, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Celli, J.; de Chastellier, C.; Franchini, D.-M.; Pizarro-Cerda, J.; Moreno, E.; Gorvel, J.-P. Brucella Evades Macrophage killing via VirB-dependent sustained interactions with the endoplasmic reticulum. J. Exp. Med. 2003, 198, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Mayorga, L.S.; Bertini, F.; Stahl, P.D. Fusion of newly formed phagosomes with endosomes in intact cells and in a cell-free system. J. Biol. Chem. 1991, 266, 6511–6517. [Google Scholar] [PubMed]
- Desjardins, M.; Celis, J.E.; van Meer, G.; Dieplinger, H.; Jahraus, A.; Griffiths, G.; Huber, L.A. Molecular characterization of phagosomes. J. Biol. Chem. 1994, 269, 32194–32200. [Google Scholar] [PubMed]
- Starr, T.; Ng, T.W.; Wehrly, T.D.; Knodler, L.A.; Celli, J. Brucella intracellular replication requires trafficking through the late endosomal/lysosomal compartment. Traffic 2008, 9, 678–694. [Google Scholar] [CrossRef] [PubMed]
- Sola-Landa, A.; Pizarro-Cerdá, J.; Grilló, M.J.; Moreno, E.; Moriyón, I.; Blasco, J.M.; Gorvel, J.P.; López-Goñi, I. A two-component regulatory system playing a critical role in plant pathogens and endosymbionts is present in Brucella abortus and controls cell invasion and virulence. Mol. Microbiol. 1998, 29, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Verri, C.; Manterola, L.; Sola-Landa, A.; Parra, A.; Cloeckaert, A.; Garin, J.; Gorvel, J.-P.; Moriyon, I.; Moreno, E.; Lopez-Goni, I. The two-component system BvrR/BvrS essential for Brucella abortus virulence regulates the expression of outer membrane proteins with counterparts in members of the Rhizobiaceae. Proc. Natl. Acad. Sci. USA 2002, 99, 12375–12380. [Google Scholar] [CrossRef] [PubMed]
- Lamontagne, J.; Butler, H.; Chaves-Olarte, E.; Hunter, J.; Schirm, M.; Paquet, C.; Tian, M.; Kearney, P.; Hamaidi, L.; Chelsky, D.; et al. Extensive cell envelope modulation is associated with virulence in Brucella abortus. J. Proteome Res. 2007, 6, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Viadas, C.; Rodríguez, M.C.; Sangari, F.J.; Gorvel, J.-P.; García-Lobo, J.M.; López-Goñi, I. Transcriptome analysis of the Brucella abortus BvrR/BvrS Two-Component Regulatory System. PLoS ONE 2010, 5, e10216. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Nunez, C.; Altamirano-Silva, P.; Alvarado-Guillen, F.; Moreno, E.; Guzman-Verri, C.; Chaves-Olarte, E. The Two-Component System BvrR/BvrS regulates the expression of the type IV Secretion System VirB in Brucella abortus. J. Bacteriol. 2010, 192, 5603–5608. [Google Scholar] [CrossRef] [PubMed]
- Boschiroli, M.L.; Ouahrani-Bettache, S.; Foulongne, V.; Michaux-Charachon, S.; Bourg, G.; Allardet-Servent, A.; Cazevieille, C.; Liautard, J.P.; Ramuz, M.; O’Callaghan, D. The Brucella suis virB operon is induced intracellularly in macrophages. Proc. Natl. Acad. Sci. USA 2002, 99, 1544–1549. [Google Scholar] [CrossRef]
- Altamirano-Silva, P.; Meza-Torres, J.; Castillo-Zeledón, A.; Ruiz-Villalobos, N.; Zuñiga-Pereira, A.M.; Chacón-Díaz, C.; Moreno, E.; Guzmán-Verri, C.; Chaves-Olarte, E. Brucella abortus senses the intracellular environment through the two-component system BvrR/BvrS allowing the adaptation to its replicative niche. Infect. Immun. 2018, 86, e00713-17. [Google Scholar] [CrossRef]
- Martínez de Tejada, G.; Pizarro-Cerdá, J.; Moreno, E.; Moriyón, I. The outer membranes of Brucella spp. are resistant to bactericidal cationic peptides. Infect. Immun. 1995, 63, 3054–3061. [Google Scholar]
- Freer, E.; Moreno, E.; Moriyón, I.; Pizarro-Cerdá, J.; Weintraub, A.; Gorvel, J.P. Brucella-Salmonella lipopolysaccharide chimeras are less permeable to hydrophobic probes and more sensitive to cationic peptides and EDTA than are their native Brucella sp. counterparts. J. Bacteriol. 1996, 178, 5867–5876. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. Protein structure and function prediction using I-TASSER. Curr. Protoc. Bioinform. 2015, 52, 5–8. [Google Scholar]
- Xu, J.; Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics 2010, 26, 889–895. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. Protein–ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. BioLiP: A semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res. 2013, 41, D1096–D1103. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Yang, J.; Zhang, Y. COFACTOR: An accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 2012, 40, W471–W477. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Freddolino, P.L.; Zhang, Y. COFACTOR: Improved protein function prediction by combining structure, sequence and protein–protein interaction information. Nucleic Acids Res. 2017, 45, W291–W299. [Google Scholar] [CrossRef]
- Dawson, N.L.; Lewis, T.E.; Das, S.; Lees, J.G.; Lee, D.; Ashford, P.; Orengo, C.A.; Sillitoe, I. CATH: An expanded resource to predict protein function through structure and sequence. Nucleic Acids Res. 2017, 45, D289–D295. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. IUCr PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Rullmannn, J.A.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Bowie, J.U.; Lüthy, R.; Eisenberg, D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991, 253, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Pontius, J.; Richelle, J.; Wodak, S.J. Deviations from standard atomic volumes as a quality measure for protein crystal structures. J. Mol. Biol. 1996, 264, 121–136. [Google Scholar] [CrossRef]
- Yan, Y.; Wen, Z.; Wang, X.; Huang, S.-Y. Addressing recent docking challenges: A hybrid strategy to integrate template-based and free protein-protein docking. Proteins Struct. Funct. Bioinform. 2017, 85, 497–512. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.-Y. HDOCK: A web server for protein–protein and protein–DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef]
- Huang, S.-Y.; Zou, X. A knowledge-based scoring function for protein-RNA interactions derived from a statistical mechanics-based iterative method. Nucleic Acids Res. 2014, 42, e55. [Google Scholar] [CrossRef]
- Huang, S.-Y.; Zou, X. An iterative knowledge-based scoring function for protein-protein recognition. Proteins Struct. Funct. Bioinform. 2008, 72, 557–579. [Google Scholar] [CrossRef]
- Yan, Y.; Huang, S. A new pairwise shape-based scoring function to consider long-range interactions for protein-protein docking. Biophys. J. 2017, 112, 470a. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- National Center of Biotechnology Information (NCBI). Available online: https://www.ncbi.nlm.nih.gov/protein/AAC28777.1?report=genbank&log$=protalign&blast_rank=24&RID=WZ0WJA2M014 (accessed on 7 December 2018).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, M.; Bonvin, A.M.J.J. 3D-DART: A DNA structure modelling server. Nucleic Acids Res. 2009, 37, W235–W239. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.; Rouchka, E.C.; Lawrence, C.E. Gibbs Recursive Sampler: Finding transcription factor binding sites. Nucleic Acids Res. 2003, 31, 3580–3585. [Google Scholar] [CrossRef] [PubMed]
- Newberg, L.A.; Thompson, W.A.; Conlan, S.; Smith, T.M.; McCue, L.A.; Lawrence, C.E. A phylogenetic Gibbs sampler that yields centroid solutions for cis-regulatory site prediction. Bioinformatics 2007, 23, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.A.; Newberg, L.A.; Conlan, S.; McCue, L.A.; Lawrence, C.E. The Gibbs Centroid Sampler. Nucleic Acids Res. 2007, 35, W232–W237. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.E.; Gullingsrud, J.; Schulten, K. A system for interactive molecular dynamics simulation. In Proceedings of the 2001 Symposium on Interactive 3D Graphics, Chapel Hill, NC, USA, 26–29 March 2001; ACM: New York, NY, USA, 2001; pp. 191–194. [Google Scholar]
- Eargle, J.; Wright, D.; Luthey-Schulten, Z. Multiple alignment of protein structures and sequences for VMD. Bioinformatics 2006, 22, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Langevine, P. On the theory of Brownian motion. Comptes Rendus Acad. Bulg. des Sci. 1908, 146, 530–533. [Google Scholar]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Vasmatzis, G.; Cornette, J.L.; DeLisi, C. Determination of atomic desolvation energies from the structures of crystallized proteins 1 1Edited by B. Honig. J. Mol. Biol. 1997, 267, 707–726. [Google Scholar] [CrossRef] [PubMed]
- Vitale, S.G.; Valenti, G.; Rapisarda, A.M.C.; Cali, I.; Marilli, I.; Zigarelli, M.; Sarpietro, G.; Cianci, A. P16INK4a as a progression/regression tumour marker in LSIL cervix lesions: Our clinical experience. Eur. J. Gynaecol. Oncol. 2016, 37, 685–688. [Google Scholar] [PubMed]
Sample Availability: Not available. |










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramírez-González, E.A.; Moreno-Lafont, M.C.; Méndez-Tenorio, A.; Cancino-Díaz, M.E.; Estrada-García, I.; López-Santiago, R. Prediction of Structure and Molecular Interaction with DNA of BvrR, a Virulence-Associated Regulatory Protein of Brucella. Molecules 2019, 24, 3137. https://doi.org/10.3390/molecules24173137
Ramírez-González EA, Moreno-Lafont MC, Méndez-Tenorio A, Cancino-Díaz ME, Estrada-García I, López-Santiago R. Prediction of Structure and Molecular Interaction with DNA of BvrR, a Virulence-Associated Regulatory Protein of Brucella. Molecules. 2019; 24(17):3137. https://doi.org/10.3390/molecules24173137
Chicago/Turabian StyleRamírez-González, Edgar A., Martha C. Moreno-Lafont, Alfonso Méndez-Tenorio, Mario E. Cancino-Díaz, Iris Estrada-García, and Rubén López-Santiago. 2019. "Prediction of Structure and Molecular Interaction with DNA of BvrR, a Virulence-Associated Regulatory Protein of Brucella" Molecules 24, no. 17: 3137. https://doi.org/10.3390/molecules24173137
APA StyleRamírez-González, E. A., Moreno-Lafont, M. C., Méndez-Tenorio, A., Cancino-Díaz, M. E., Estrada-García, I., & López-Santiago, R. (2019). Prediction of Structure and Molecular Interaction with DNA of BvrR, a Virulence-Associated Regulatory Protein of Brucella. Molecules, 24(17), 3137. https://doi.org/10.3390/molecules24173137