
The Role of Active-Site Residues Phe98, His239, and Arg243 in DNA Binding and in the Catalysis of Human Uracil–DNA Glycosylase SMUG1

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
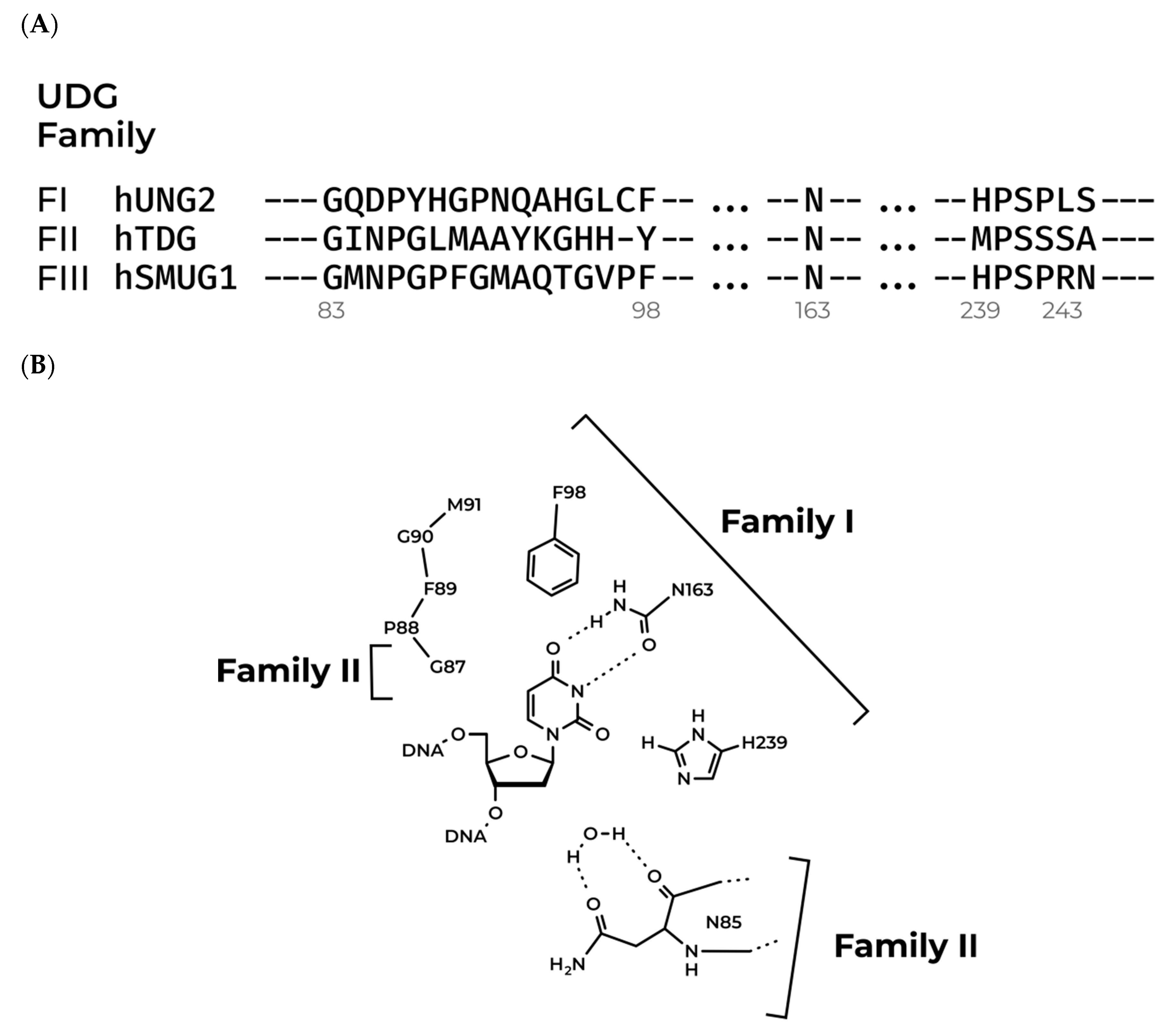
2.1. Model Structure of hSMUG1
2.2. Modeling the Structure of the Enzyme–DNA Complex
2.3. Protein Expression and Purification
2.4. Oligodeoxynucleotides (ODNs)
2.5. PAGE Analysis of Time-Course Experiments
2.6. Circular Dichroism (CD) Spectra
2.7. Stopped-Flow Fluorescence Kinetics
2.8. Kinetic Data Analysis
3. Results and Discussion
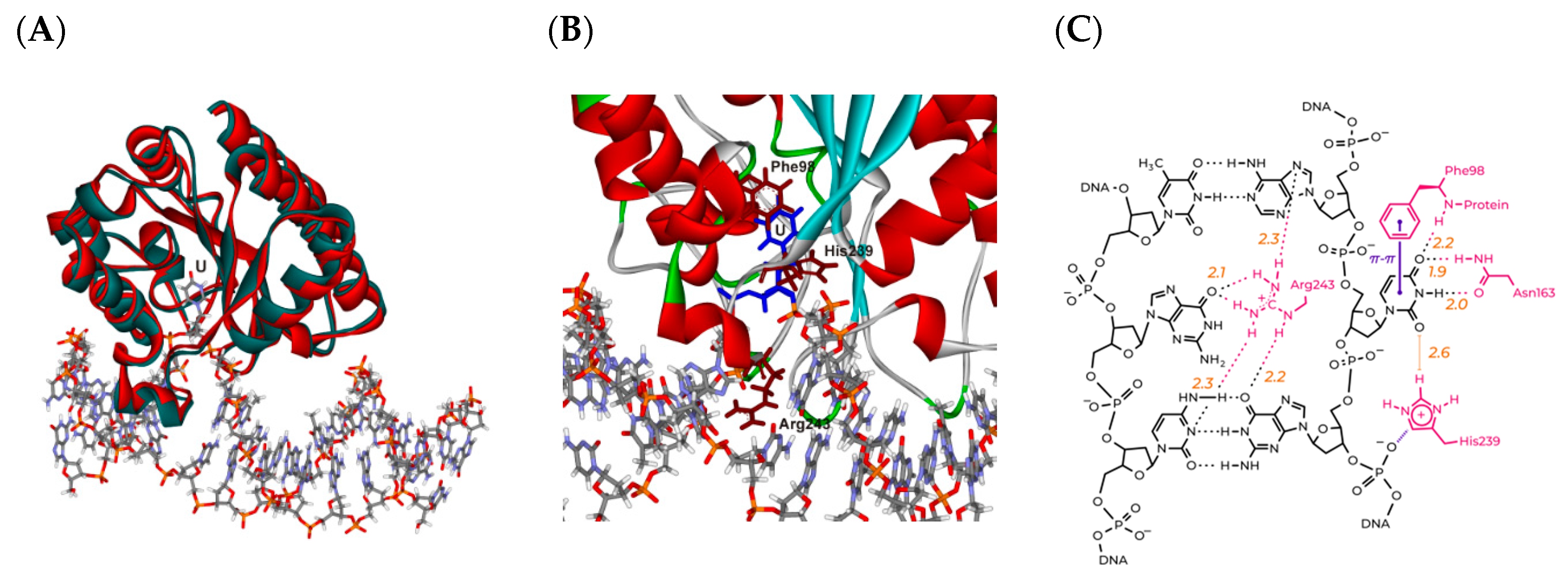
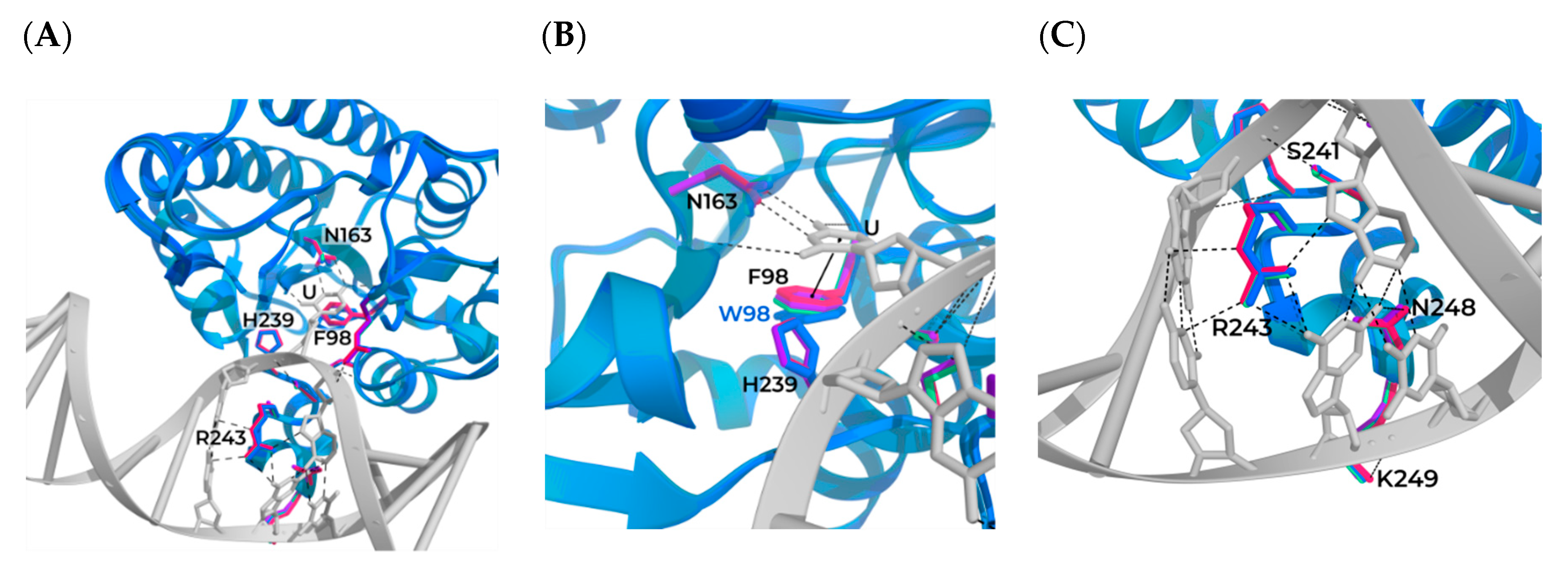
3.1. Modeling of the Structures of hSMUG1 Complexes with a DNA Substrate
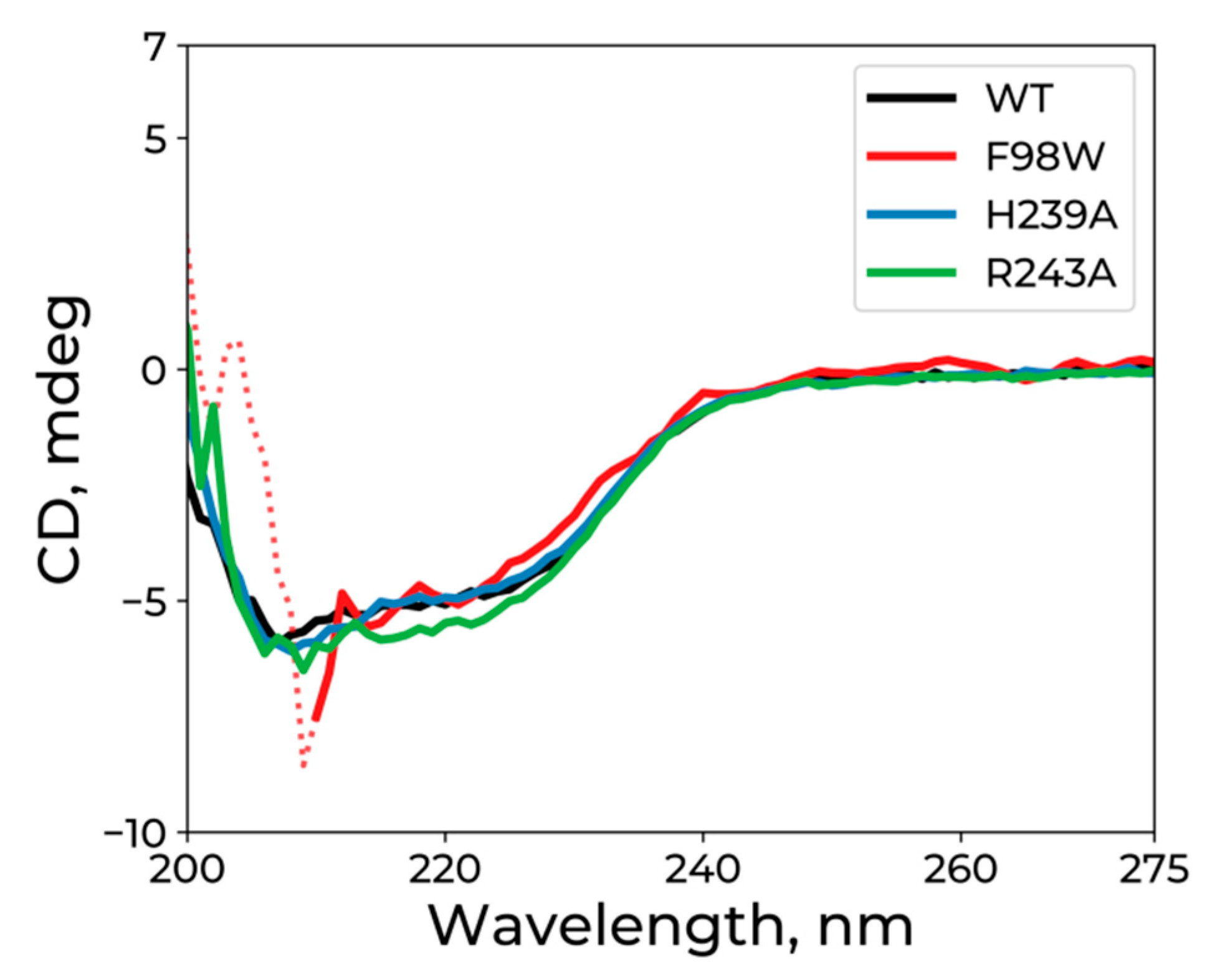
3.2. CD Analysis of WT hSMUG1 and of the Mutants
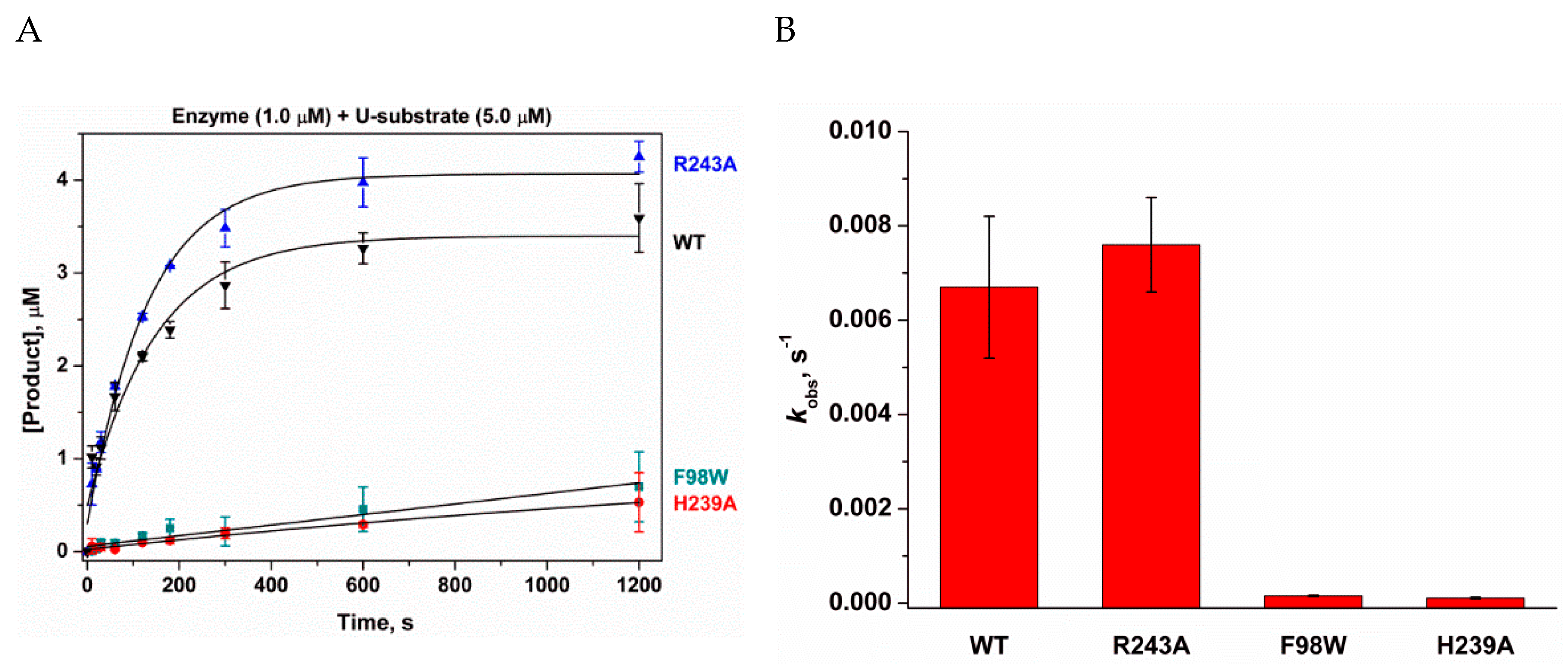
3.3. Relative N-Glycosylase Activity of WT hSMUG1 and of the Mutants
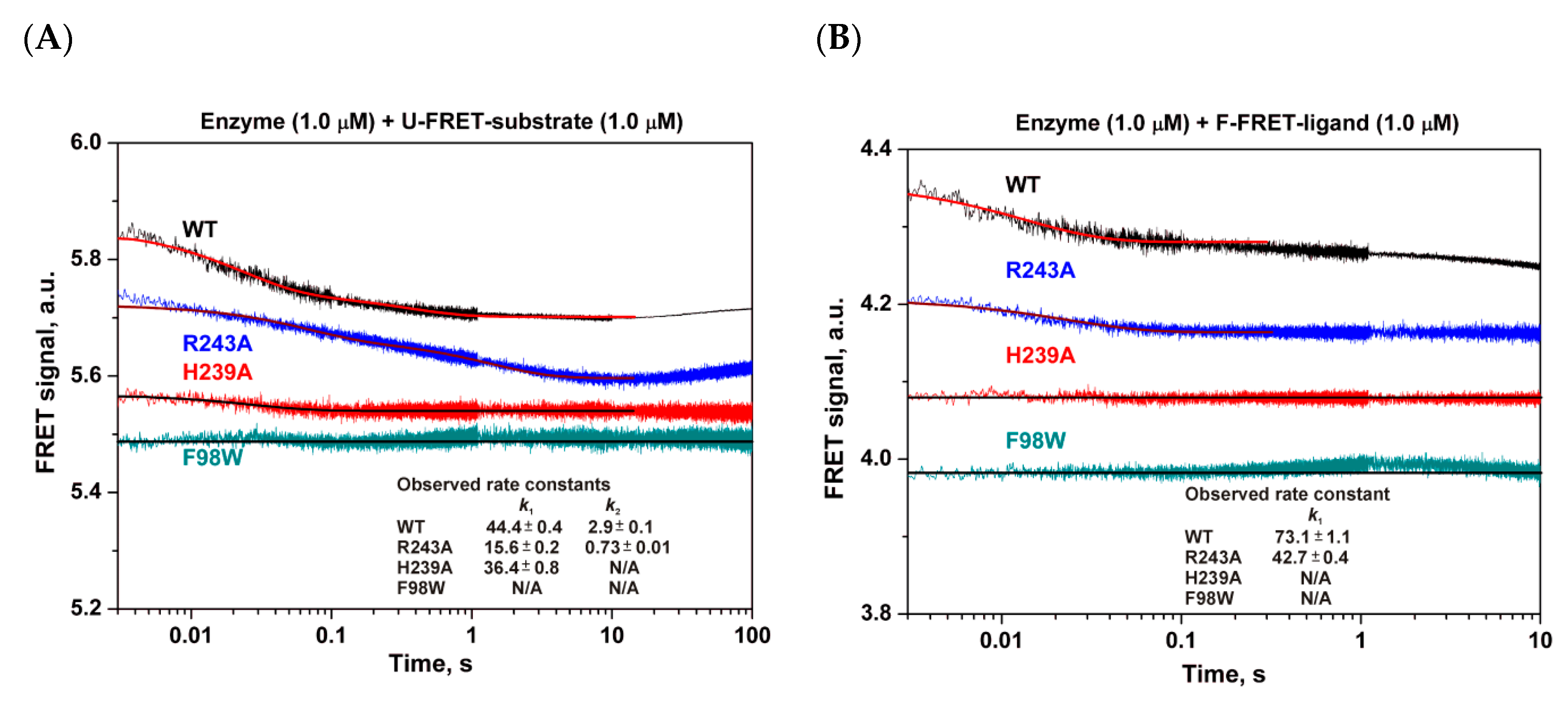
3.4. Pre-Steady-State Analysis of Conformational Dynamics of WT hSMUG1 and of the Mutants
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| SMUG1 | single-stranded selective monofunctional uracil–DNA glycosylase; |
| AP-site | apurinic/apyrimidinic site; |
| F-site | (2R,3S)-2-(hydroxymethyl)-3-hydroxytetrahydrofuran residue; |
| ODN | oligodeoxyribonucleotide; |
| PAGE | polyacrylamide gel electrophoresis; |
| BER | base excision repair; |
| FRET | Förster resonance energy transfer; |
| AMBER | Assisted Model Building with Energy Refinement. |
References
- Kavli, B.; Sundheim, O.; Akbari, M.; Otterlei, M.; Nilsen, H.; Skorpen, F.; Aas, P.A.; Hagen, L.; Krokan, H.E.; Slupphaug, G. hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J. Biol. Chem. 2002, 277, 39926–39936. [Google Scholar] [CrossRef]
- Wibley, J.E.A.; Waters, T.R.; Haushalter, K.; Verdine, G.L.; Pearl, L.H. Structure and specificity of the vertebrate anti-mutator uracil-DNA glycosylase SMUG1. Mol. Cell 2003, 11, 1647–1659. [Google Scholar] [CrossRef]
- Hashimoto, H.; Hong, S.; Bhagwat, A.S.; Zhang, X.; Cheng, X. Excision of 5-hydroxymethyluracil and 5-carboxylcytosine by the thymine DNA glycosylase domain: Its structural basis and implications for active DNA demethylation. Nucleic Acids Res. 2012, 40, 10203–10214. [Google Scholar] [CrossRef]
- Hashimoto, H.; Zhang, X.; Cheng, X. Excision of thymine and 5-hydroxymethyluracil by the MBD4 DNA glycosylase domain: Structural basis and implications for active DNA demethylation. Nucleic Acids Res. 2012, 40, 8276–8284. [Google Scholar] [CrossRef]
- Sjolund, A.B.; Senejani, A.G.; Sweasy, J.B. MBD4 and TDG: Multifaceted DNA glycosylases with ever expanding biological roles. Mutat. Res. 2013, 743–744, 12–25. [Google Scholar] [CrossRef]
- Ito, S.; Kuraoka, I. Epigenetic modifications in DNA could mimic oxidative DNA damage: A double-edged sword. DNA Repair 2015, 32, 52–57. [Google Scholar] [CrossRef]
- Schormann, N.; Ricciardi, R.; Chattopadhyay, D. Uracil-DNA glycosylases—Structural and functional perspectives on an essential family of DNA repair enzymes. Protein Sci. 2014, 23, 1667–1685. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, Z.; Crombet, L.; Amaya, M.F.; Liu, Y.; Zhang, X.; Kuang, W.; Ma, P.; Niu, L.; Qi, C. Crystal structure of the mismatch-specific thymine glycosylase domain of human methyl-CpG-binding protein MBD4. Biochem. Biophys. Res. Commun. 2011, 412, 425–428. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Iakovlev, D.A.; Misovets, I.V.; Ishchenko, A.A.; Saparbaev, M.K.; Kuznetsov, N.A.; Fedorova, O.S. Pre-steady-state kinetic analysis of damage recognition by human single-strand selective monofunctional uracil-DNA glycosylase SMUG1. Mol. Biosyst. 2017, 13, 2638–2649. [Google Scholar] [CrossRef]
- Zhang, Z.; Shen, J.; Yang, Y.; Li, J.; Cao, W.; Xie, W. Structural basis of substrate specificity in geobacter metallireducens SMUG1. ACS Chem. Biol. 2016, 11, 1729–1736. [Google Scholar] [CrossRef]
- Matsubara, M.; Tanaka, T.; Terato, H.; Ohmae, E.; Izumi, S.; Katayanagi, K.; Ide, H. Mutational analysis of the damage-recognition and catalytic mechanism of human SMUG1 DNA glycosylase. Nucleic Acids Res. 2004, 32, 5291–5302. [Google Scholar] [CrossRef]
- Nelson, S.R.; Dunn, A.R.; Kathe, S.D.; Warshaw, D.M.; Wallace, S.S. Two glycosylase families diffusively scan DNA using a wedge residue to probe for and identify oxidatively damaged bases. Proc. Natl. Acad. Sci. USA 2014, 111, E2091–E2099. [Google Scholar] [CrossRef]
- Lee, A.J.; Wallace, S.S. Hide and seek: How do DNA glycosylases locate oxidatively damaged DNA bases amidst a sea of undamaged bases? Free Radic. Biol. Med. 2017, 107, 170–178. [Google Scholar] [CrossRef]
- Kladova, O.A.; Kuznetsova, A.A.; Fedorova, O.S.; Kuznetsov, N.A. Mutational and kinetic analysis of lesion recognition by escherichia coli endonuclease VIII. Genes 2017, 8, 140. [Google Scholar] [CrossRef]
- Kuznetsov, N.A.; Kladova, O.A.; Kuznetsova, A.A.; Ishchenko, A.A.; Saparbaev, M.K.; Zharkov, D.O.; Fedorova, O.S. Conformational dynamics of DNA repair by escherichia coli endonuclease III. J. Biol. Chem. 2015, 290, 14338–14349. [Google Scholar] [CrossRef]
- Kuznetsov, N.A.; Bergonzo, C.; Campbell, A.J.; Li, H.; Mechetin, G.V.; de los Santos, C.; Grollman, A.P.; Fedorova, O.S.; Zharkov, D.O.; Simmerling, C. Active destabilization of base pairs by a DNA glycosylase wedge initiates damage recognition. Nucleic Acids Res. 2015, 43, 272–281. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Kuznetsov, N.A.; Ishchenko, A.A.; Saparbaev, M.K.; Fedorova, O.S. Step-by-step mechanism of DNA damage recognition by human 8-oxoguanine DNA glycosylase. Biochim. Biophys. Acta 2014, 1840, 387–395. [Google Scholar] [CrossRef]
- Shapovalov, M.V.; Dunbrack, R.L. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation all atom force field for the simulation of proteins, nucleic acids and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Popov, A.V.; Vorob’ev, Y.N. GUI-BioPASED: A program for molecular dynamics simulations of biopolymers with a graphical user interface. Mol. Biol. 2010, 44, 735–742. [Google Scholar] [CrossRef]
- Lazaridis, T.; Karplus, M. Effective energy function for proteins in solution. Proteins 1999, 35, 133–152. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Yakovlev, D.A.; Kuznetsova, A.A.; Fedorova, O.S.; Kuznetsov, N.A. Search for modified DNA sites with the human methyl-CpG-binding enzyme MBD4. Acta Nat. 2017, 9, 88–98. [Google Scholar] [CrossRef]
- Kuznetsov, N.A.; Kiryutin, A.S.; Kuznetsova, A.A.; Panov, M.S.; Barsukova, M.O.; Yurkovskaya, A.V.; Fedorova, O.S. The formation of catalytically competent enzyme-substrate complex is not a bottleneck in lesion excision by human alkyladenine DNA glycosylase. J. Biomol. Struct. Dyn. 2017, 35, 950–967. [Google Scholar] [CrossRef]
- Miroshnikova, A.D.; Kuznetsova, A.A.; Vorobjev, Y.N.; Kuznetsov, N.A.; Fedorova, O.S. Effects of mono- and divalent metal ions on DNA binding and catalysis of human apurinic/apyrimidinic endonuclease 1. Mol. BioSyst. 2016, 12, 1527–1539. [Google Scholar] [CrossRef]
- Kladova, O.A.; Kuznetsov, N.A.; Fedorova, O.S. Thermodynamics of the DNA repair process by endonuclease VIII. Acta Nat. 2019, 11, 29–37. [Google Scholar] [CrossRef]
- Pettersen, H.S.; Sundheim, O.; Gilljam, K.M.; Slupphaug, G.; Krokan, H.E.; Kavli, B. Uracil-DNA glycosylases SMUG1 and UNG2 coordinate the initial steps of base excision repair by distinct mechanisms. Nucleic Acids Res. 2007, 35, 3879–3892. [Google Scholar] [CrossRef]
Sample Availability: Samples of the F98W, H239A, and R243A hSMUG1are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Shorthand | Sequence |
|---|---|
| U-substrate | 5′-GCTCAUGTACAGAGCTG-3′ 3′-CGAGTGCATGTCTCGAC-5′ |
| F-ligand | 5′-GCTCAFGTACAGAGCTG-3′ 3′-CGAGTGCATGTCTCGAC-5′ |
| U-FRET-substrate | 5′-FAM-GCTCAUGTACAGAGCTG-3′ 3′-CGAGTGCATGTCTCGAC-BHQ1-5′ |
| F-FRET-ligand | 5′-FAM-GCTCAFGTACAGAGCTG-3′ 3′-CGAGTGCATGTCTCGAC-BHQ1-5′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iakovlev, D.A.; Alekseeva, I.V.; Vorobjev, Y.N.; Kuznetsov, N.A.; Fedorova, O.S. The Role of Active-Site Residues Phe98, His239, and Arg243 in DNA Binding and in the Catalysis of Human Uracil–DNA Glycosylase SMUG1. Molecules 2019, 24, 3133. https://doi.org/10.3390/molecules24173133
Iakovlev DA, Alekseeva IV, Vorobjev YN, Kuznetsov NA, Fedorova OS. The Role of Active-Site Residues Phe98, His239, and Arg243 in DNA Binding and in the Catalysis of Human Uracil–DNA Glycosylase SMUG1. Molecules. 2019; 24(17):3133. https://doi.org/10.3390/molecules24173133
Chicago/Turabian StyleIakovlev, Danila A., Irina V. Alekseeva, Yury N. Vorobjev, Nikita A. Kuznetsov, and Olga S. Fedorova. 2019. "The Role of Active-Site Residues Phe98, His239, and Arg243 in DNA Binding and in the Catalysis of Human Uracil–DNA Glycosylase SMUG1" Molecules 24, no. 17: 3133. https://doi.org/10.3390/molecules24173133
APA StyleIakovlev, D. A., Alekseeva, I. V., Vorobjev, Y. N., Kuznetsov, N. A., & Fedorova, O. S. (2019). The Role of Active-Site Residues Phe98, His239, and Arg243 in DNA Binding and in the Catalysis of Human Uracil–DNA Glycosylase SMUG1. Molecules, 24(17), 3133. https://doi.org/10.3390/molecules24173133