Crepidatumines C and D, Two New Indolizidine Alkaloids from Dendrobium crepidatum Lindl. ex Paxt.


 and
and
Abstract
:
1. Introduction
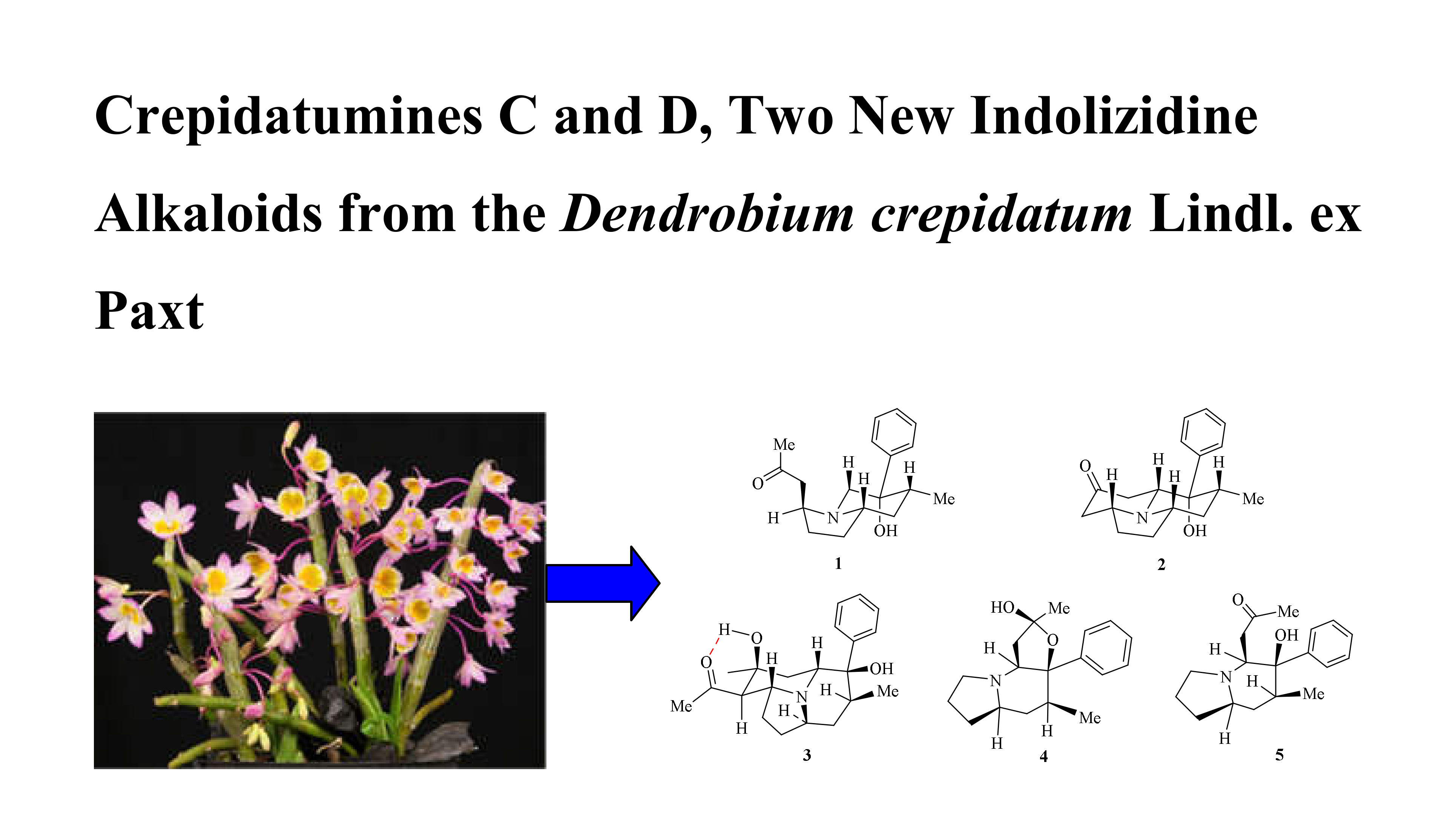
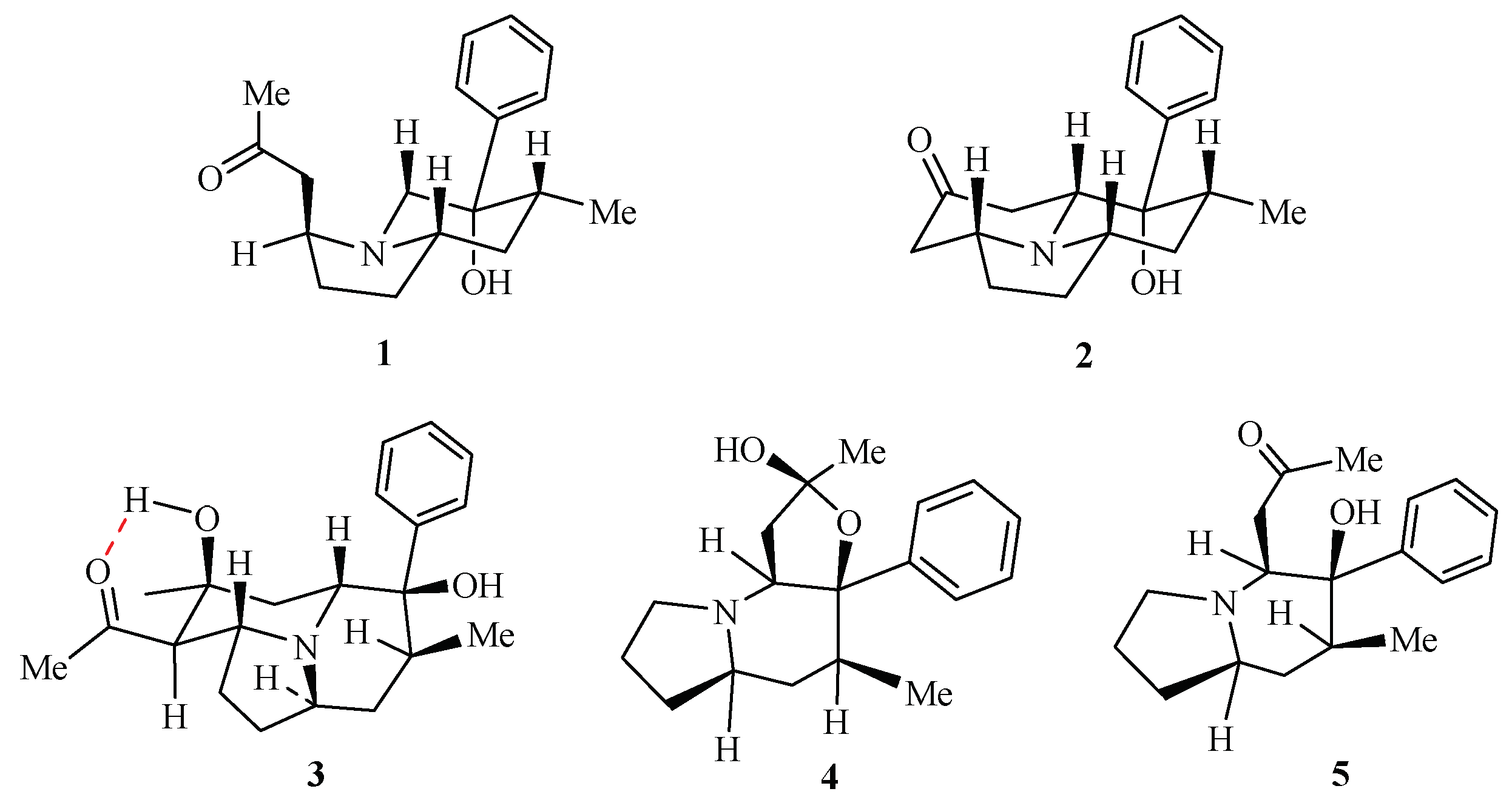
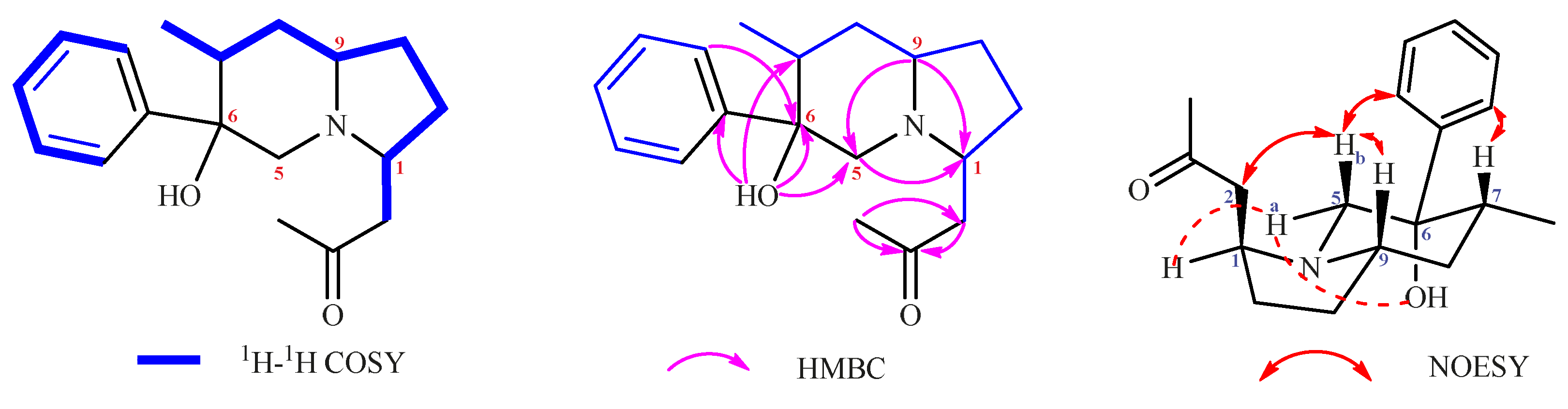
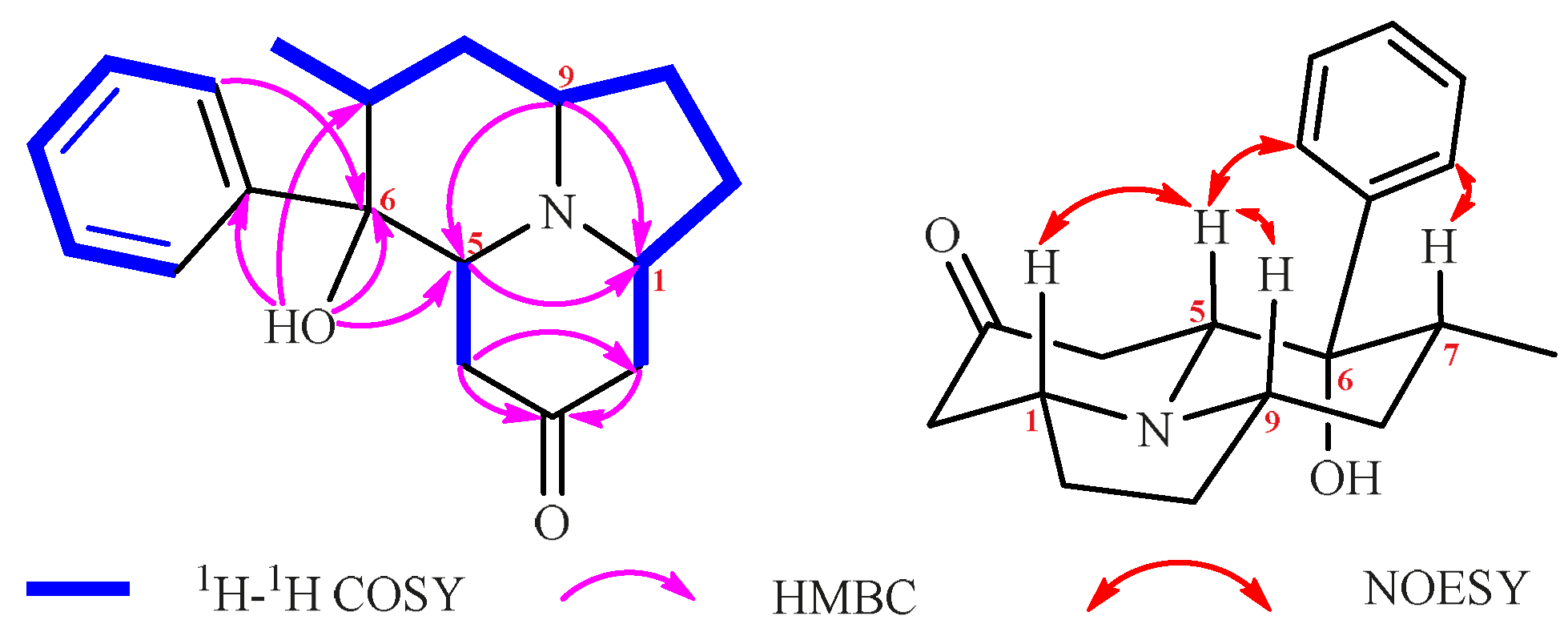
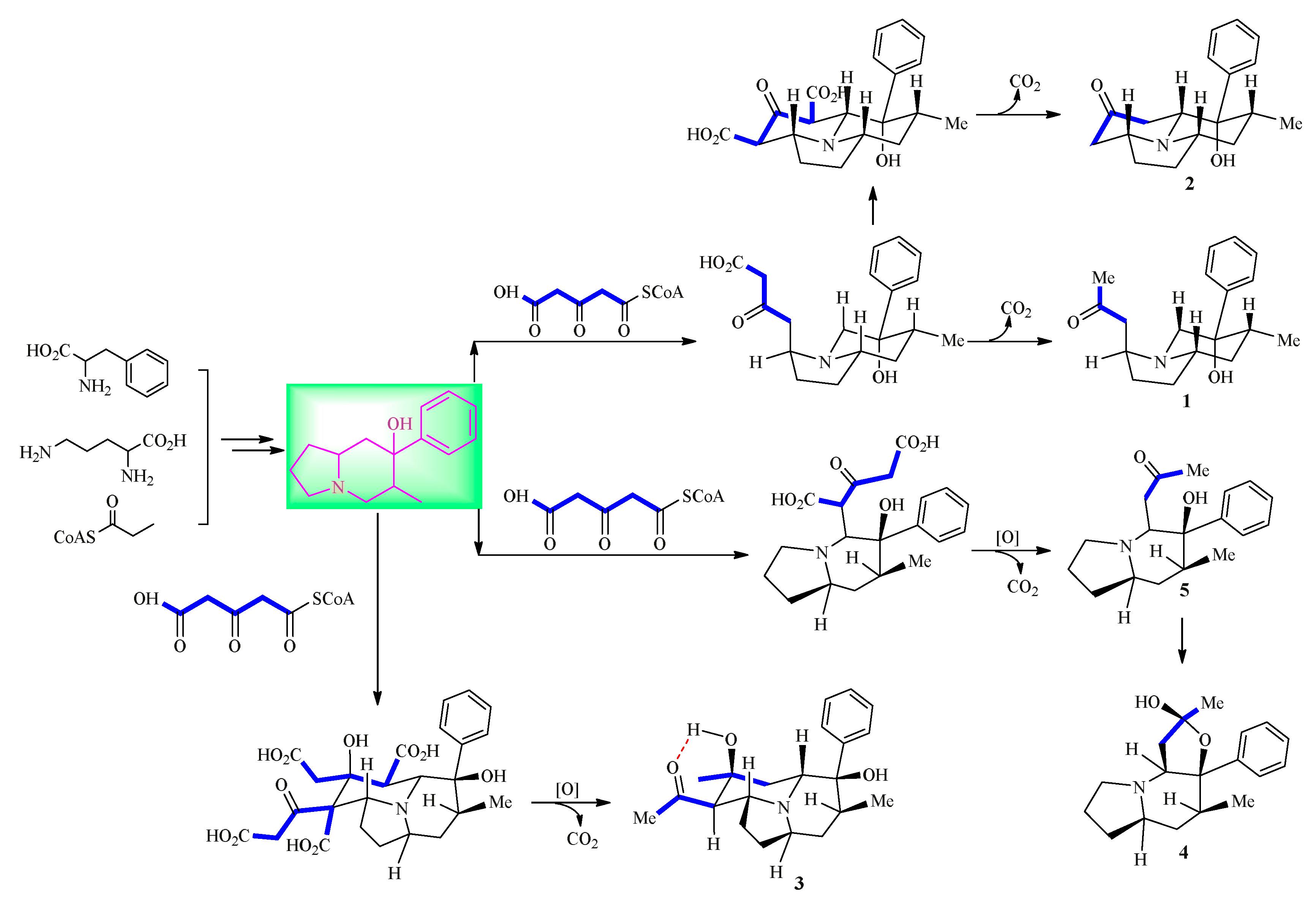
2. Results and Discussion
3. Experimental Section
3.1. General Experimental Procedures
3.2. Plant Materials
3.3. Extraction and Isolation
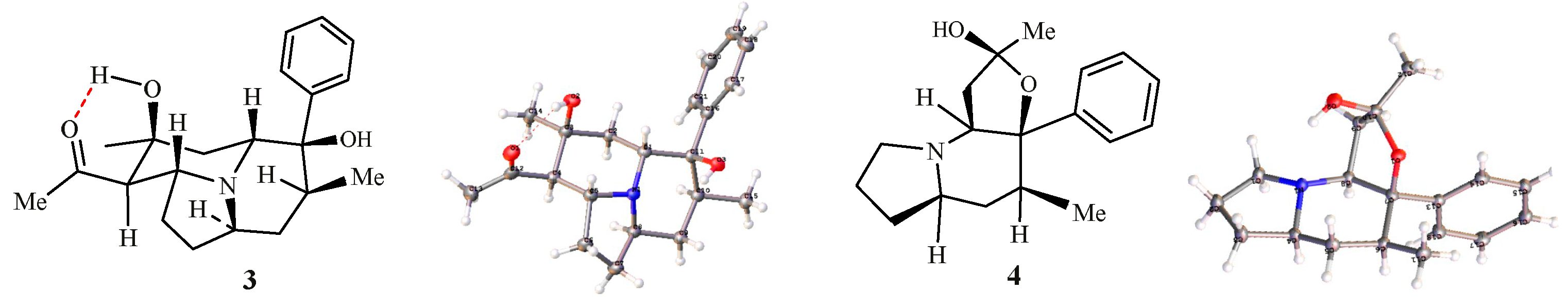
3.4. X-Ray Crystallographic Analysis of 3 and 4.
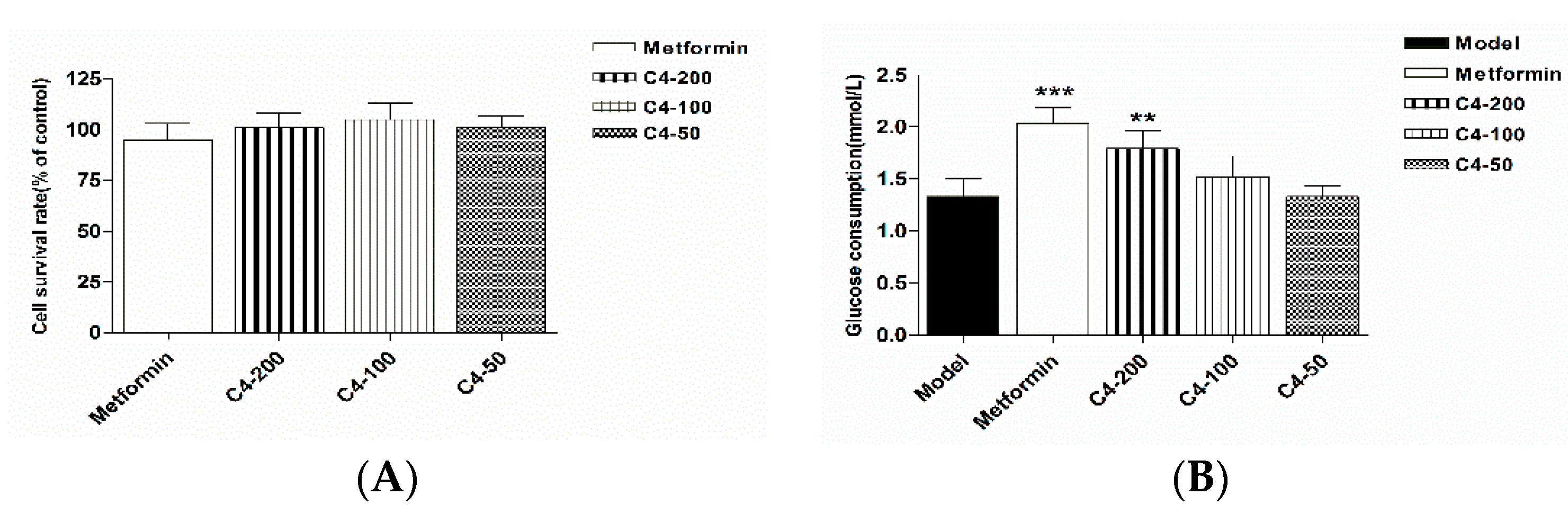
3.5. In Vitro Evaluation of Compound 4
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ng, T.B.; Liu, J.; Wong, J.H.; Ye, X.; Sze, S.C.W.; Yao, T.; Zhang, K.Y. Review of research on Dendrobium, a prized folk medicine. Appl. Microbiol. Biotechnol. 2012, 93, 1795–1803. [Google Scholar] [CrossRef]
- Wan, X. Resource and application of Dendrobium. J. Tradit. Chin. Med. 2010, 51, 957. [Google Scholar]
- Kende, A.S.; Bentley, T.J.; Mader, R.A.; Ridge, D. Simple total synthesis of (+)-dendrobine. J. Am. Chem. Soc. 1974, 96, 323–342. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhou, B.J. Chemical components of Dendrobium crepidatum and their neurite outgrowth enhancing activities. Nat. Prod. Bioprospect. 2013, 3, 70–73. [Google Scholar] [CrossRef]
- Elander, M.; Leander, K.; Rosenblom, J.; Ruusa, E. Studies on orchidaceae alkaloids. XXXII. Crepidine, crepidamine and dendrocrepine, three alkaloids from Dendrobium crepidatum Lindl. Acta Chem. Scand. 1973, 5, 1907–1913. [Google Scholar] [CrossRef]
- Hu, Y.; Ren, J.; Wang, L.; Zhao, X.; Zhang, M.; Shimizu, K.; Zhang, C. Protective effects of total alkaloids from Dendrobium crepidatum against LPS-induced acute lung injury in mice and its chemical components. Phytochemistry 2018, 149, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, C.F.; Zhao, X.; Wang, Y.; Feng, D.Q.; Zhang, M.; Xie, H.F. (±)-Homocrepidine A, a pair of anti-inflammatory enantiomeric octahydroindolizine alkaloid dimers from Dendrobium crepidatum. J. Nat. Prod. 2015, 23, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Kierkegaard, P.; Pilotti, A.; Leander, K. Studies on orchidaceae alkaloids. XX.* The constitution and relative configuration of crepidine, an alkaloid from Dendrobium crepidatum Lindl. Acta Chem. Scand. 1970, 24, 3757–3759. [Google Scholar] [CrossRef]
- Leete, E.; Robert, M. Steric inhibition of the rotation of the phenyl groups in 2,6-dimethyl-1-phenylcyclohexanol and dendrocrepine detected by means of carbon-13 nuclear magnetic resonance. Tetrahedron. Lett. 1978, 19, 5163–5166. [Google Scholar] [CrossRef]
- Xu, X.L.; Yang, Y.M.; Zhang, X.Y.; Li, Z.S.; Zhou, H.G.; Bai, Y.B.; Yu, M.; Ding, G.; Li, B. Crepidatumines A and B, two novel indolizidine alkaloids from Dendrobium crepidatum Lindl. ex Paxt. J. Nat. Prod. 2019. (submitted). [Google Scholar]
Sample Availability: Samples of the compounds (1 and 2) are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pos | 1 | 2 | ||
|---|---|---|---|---|
| δC b, Type | δH a, mult. (J in Hz) | δC b, Type | δH a, mult. (J in Hz) | |
| 1 | 74.8, CH | 4.10, m | 59.9, CH | 3.37, m |
| 2 | 48.6, CH2 | 2.46, m | 44.1, CH2 | 2.33, dd (10.8, 13.8) 1.92, ddd (1.8, 4.2, 13.8) |
| 3 | 206.8, qC | 209.6, qC | ||
| 4 | 30.9, CH3 | 2.07, s | 38.6, CH2 | 1.13, ddd (1.8, 3.0, 13.2) 2.63, t (13.2) |
| 5a 5b | 67.1, CH2 | 2.95, d (10.8) 2.69, d (10.8) | 66.7, CH | 3.00, dd (3.0, 13.2) |
| 6 | 76.5, qC | 76.2, qC | ||
| 7 | 38.4, CH | 1.96, m | 31.7, CH | 2.46, m |
| 8 | 36.3, CH2 | 1.49, m | 35.5, CH2 | 1.46, m 1.74, dt (3.0, 11.4) |
| 9 | 63.9, CH | 2.43, m | 51.4, CH | 3.15, m |
| 10 | 30.2, CH2 | 1.65, m 1.46, m | 29.7, CH2 | 1.40, m 1.97, m |
| 11 | 30.8, CH2 | 1.67, m 1.25, m | 29.1, CH2 | 1.39, m 2.04, m |
| 12 | 14.4, CH3 | 0.48, d (6.6) | 16.1, CH3 | 0.71, d (6.6) |
| 1′ | 146.1, qC | 143.9, qC | ||
| 2′/6′ | 128.2, CH | 7.46, dd (1.2, 8.4) | 126.9, CH | 7.41, br d (7.2) |
| 3′/5′ | 125.6, CH | 7.31, br d (8.4) | 128.2, CH | 7.34, dd (7.2) |
| 4′ | 126.8, CH | 7.21, br t (8.4) | 127.0, CH | 7.24, dd (7.2) |
| 6-OH | 4.76, s | 4.53, s | ||
| Identification Code | 3 |
|---|---|
| Empirical formula | C21H29NO3 |
| Formula weight | 343.45 |
| Temperature/K | 109.1(3) |
| Crystal system | orthorhombic |
| Space group | P212121 |
| a/Å, b/Å, c/Å | 5.7476(3), 17.5786(5), 17.7942(6) |
| α/°, β/°, γ/° | 90, 90, 90 |
| Volume/Å3 | 1797.84(11) |
| Z | 4 |
| ρcalc/mg mm−3 | 1.269 |
| μ/mm−1 | 0.666 |
| F (000) | 744 |
| Crystal size/mm3 | 0.35 × 0.12 × 0.02 |
| 2θ range for data collection | 7.06 to 142.74° |
| Index ranges | −6 ≤ h ≤ 7, −18 ≤ k ≤ 21, −21 ≤ l ≤ 19 |
| Reflections collected | 9652 |
| Independent reflections | 3383[R(int) = 0.0609 (inf-0.9Å)] |
| Data/restraints/parameters | 3383/0/231 |
| Goodness-of-fit on F2 | 1.030 |
| Final R indexes [I > 2σ (I) i.e., Fo > 4σ (Fo)] | R1 = 0.0510, wR2 = 0.1302 |
| Final R indexes [all data] | R1 = 0.0551, wR2 = 0.1367 |
| Largest diff. peak/hole/e Å−3 | 0.275/−0.288 |
| Flack Parameters | 0.2(2) |
| Completeness | 0.993 |
| Identification Code | 4 |
|---|---|
| Empirical formula | C20H27NO4 |
| Formula weight | 345.42 |
| Temperature/K | 107.75(10) |
| Crystal system | orthorhombic |
| Space group | P212121 |
| a/Å, b/Å, c/Å | 6.6679(4), 10.7681(4), 24.2111(9) |
| α/°, β/°, γ/° | 90, 90, 90 |
| Volume/Å3 | 1738.38(13) |
| Z | 4 |
| ρcalc/mg mm−3 | 1.320 |
| μ/mm−1 | 0.737 |
| F (000) | 744 |
| Crystal size/mm3 | 0.350 × 0.340 × 0.100 |
| 2θ range for data collection | 8.988 to 142.446° |
| Index ranges | −4 ≤ h ≤ 7, −11 ≤ k ≤ 13, −29 ≤ l ≤ 29 |
| Reflections collected | 5698 |
| Independent reflections | 3256[R(int) = 0.0271 (inf-0.9Å)] |
| Data/restraints/parameters | 3256/0/230 |
| Goodness-of-fit on F2 | 1.054 |
| Final R indexes [I > 2σ (I) i.e., Fo > 4σ (Fo)] | R1 = 0.0397, wR2 = 0.1072 |
| Final R indexes [all data] | R1 = 0.0421, wR2 = 0.1106 |
| Largest diff. peak/hole/e Å−3 | 0.312/−0.240 |
| Flack Parameters | −0.13(14) |
| Completeness | 0.9984 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.; Li, Z.; Yang, R.; Zhou, H.; Bai, Y.; Yu, M.; Ding, G.; Li, B. Crepidatumines C and D, Two New Indolizidine Alkaloids from Dendrobium crepidatum Lindl. ex Paxt. Molecules 2019, 24, 3071. https://doi.org/10.3390/molecules24173071
Xu X, Li Z, Yang R, Zhou H, Bai Y, Yu M, Ding G, Li B. Crepidatumines C and D, Two New Indolizidine Alkaloids from Dendrobium crepidatum Lindl. ex Paxt. Molecules. 2019; 24(17):3071. https://doi.org/10.3390/molecules24173071
Chicago/Turabian StyleXu, Xiaolin, Zesheng Li, Runmei Yang, Houguang Zhou, Yanbin Bai, Meng Yu, Gang Ding, and Biao Li. 2019. "Crepidatumines C and D, Two New Indolizidine Alkaloids from Dendrobium crepidatum Lindl. ex Paxt." Molecules 24, no. 17: 3071. https://doi.org/10.3390/molecules24173071
APA StyleXu, X., Li, Z., Yang, R., Zhou, H., Bai, Y., Yu, M., Ding, G., & Li, B. (2019). Crepidatumines C and D, Two New Indolizidine Alkaloids from Dendrobium crepidatum Lindl. ex Paxt. Molecules, 24(17), 3071. https://doi.org/10.3390/molecules24173071