Functionalized 1,3-Thiazolidin-4-Ones from 2-Oxo-Acenaphthoquinylidene- and [2.2]Paracyclophanylidene-Thiosemicarbazones


 , ,
, ,
Abstract
:
1. Introduction
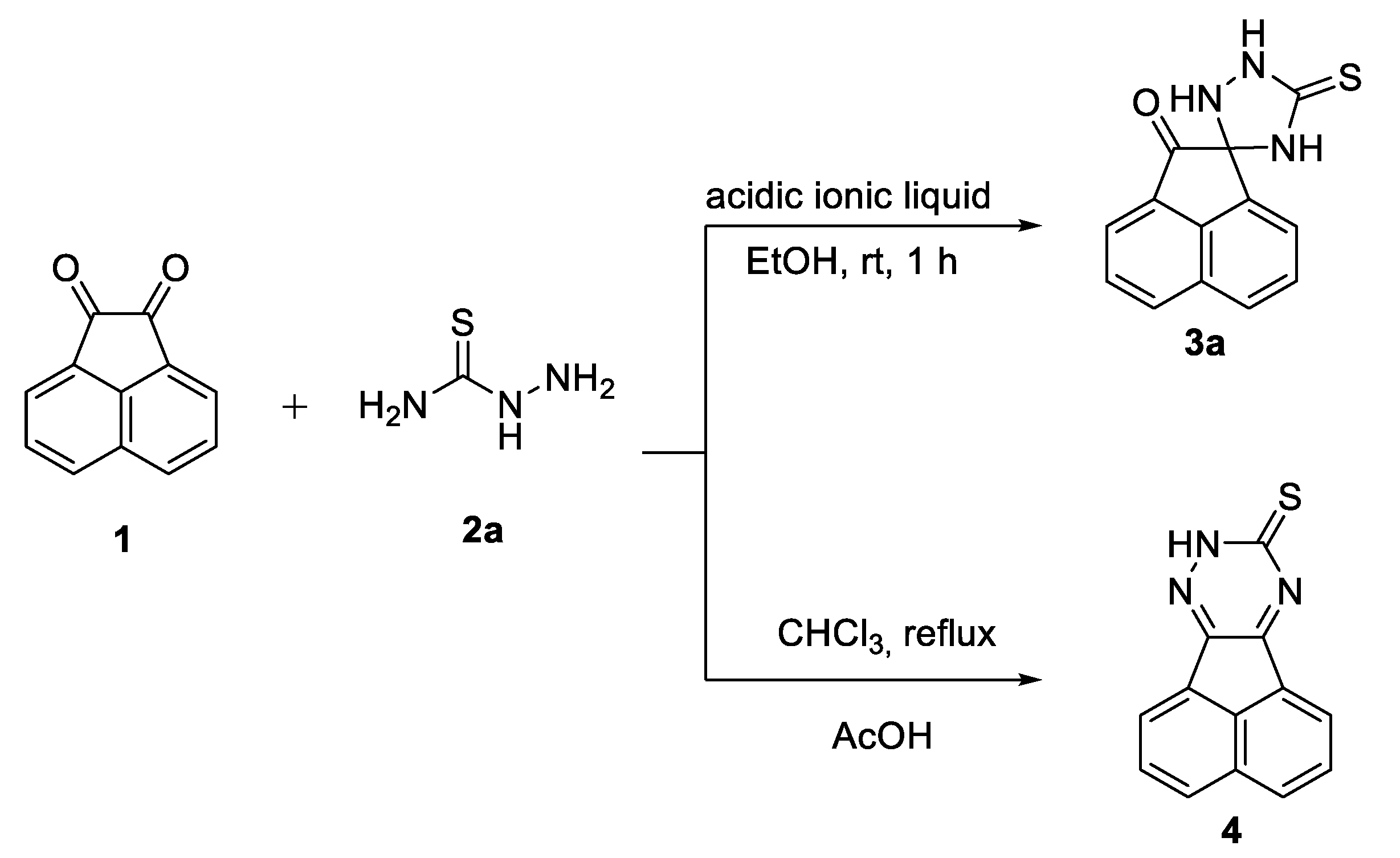
2. Results and Discussion
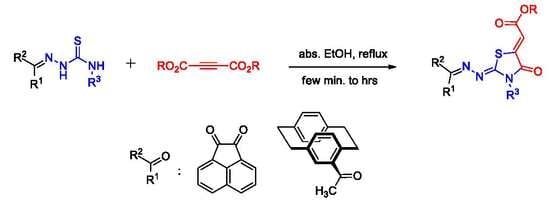
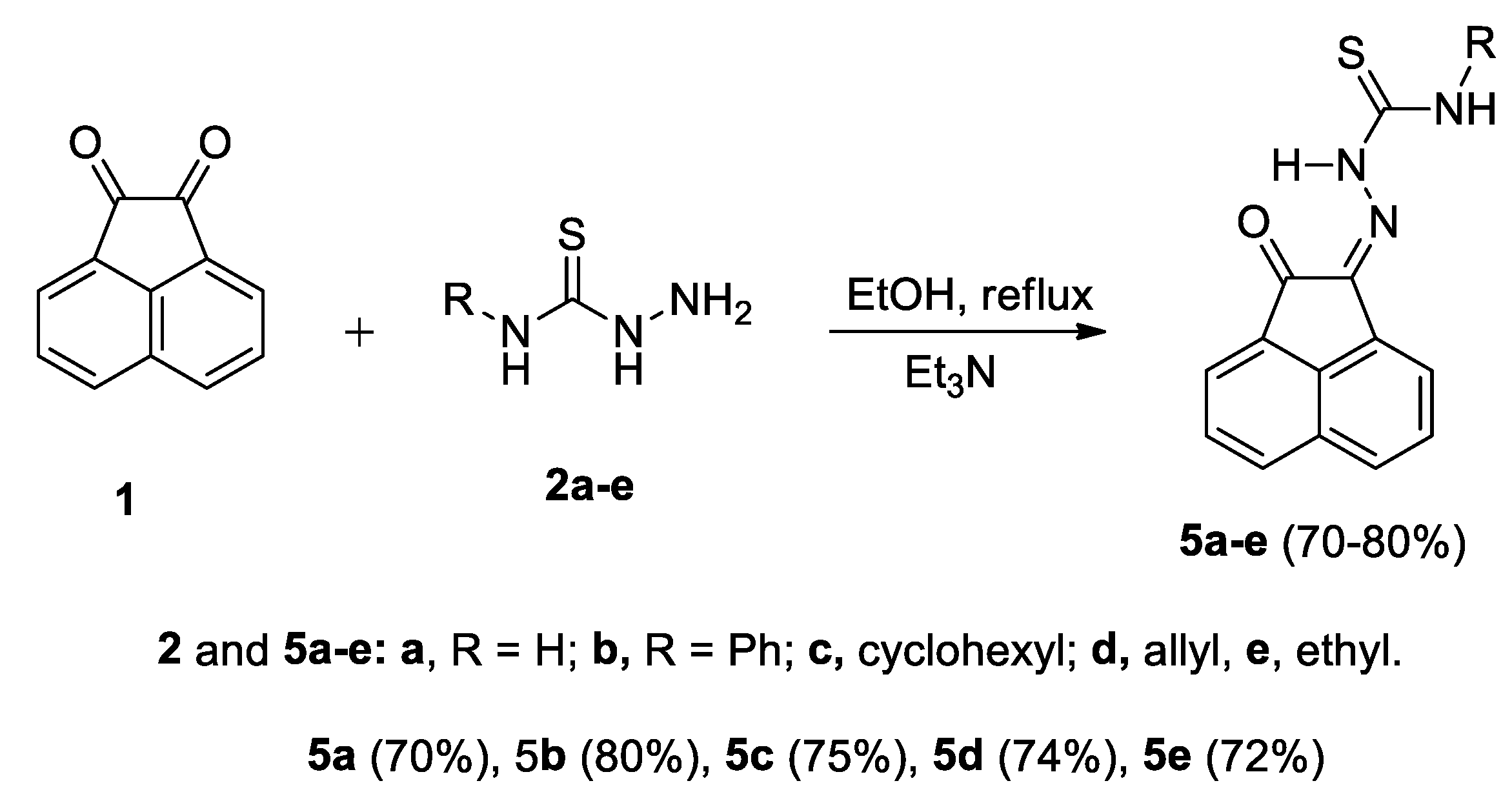
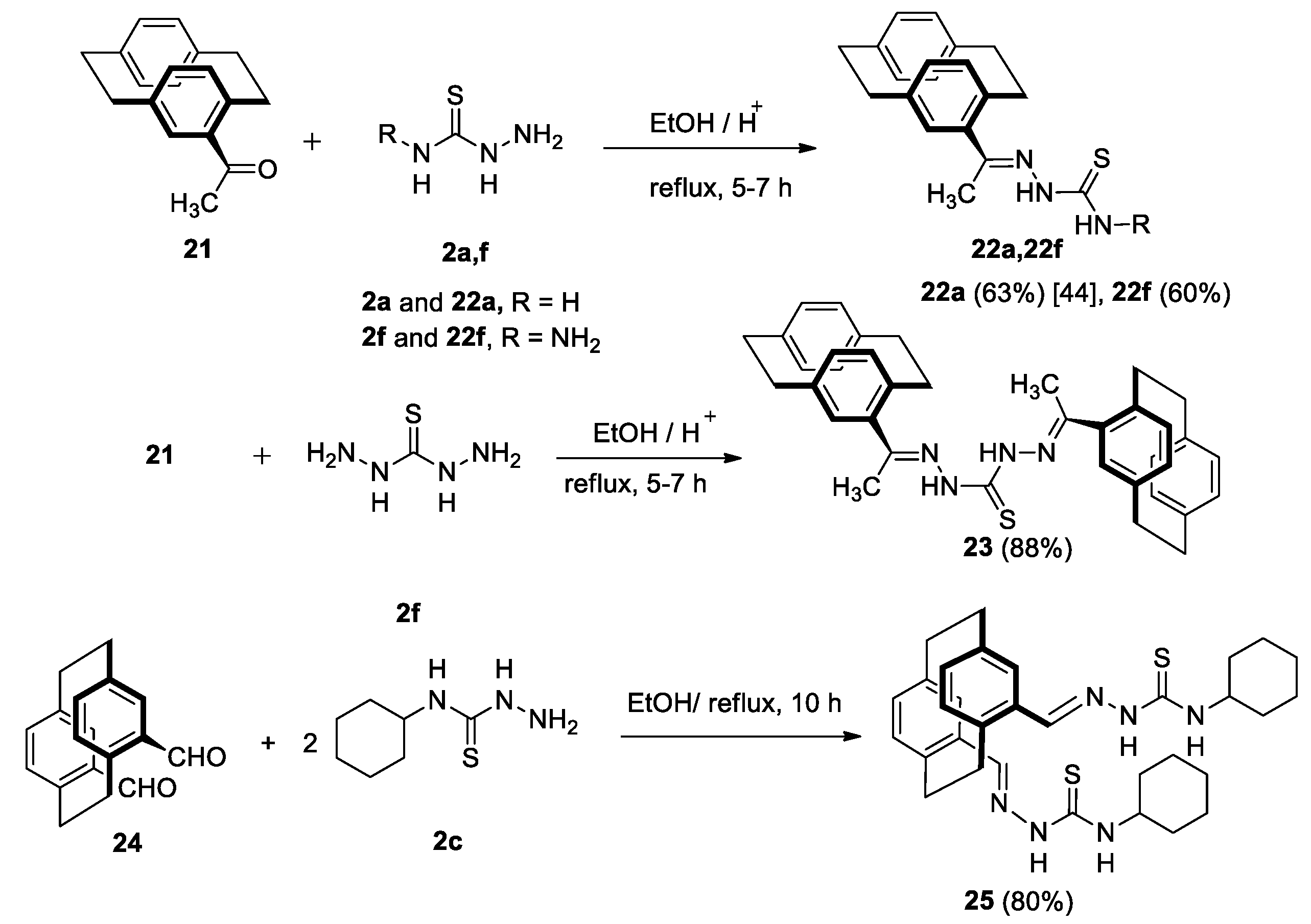
2.1. Preparation of Compounds 5a–e
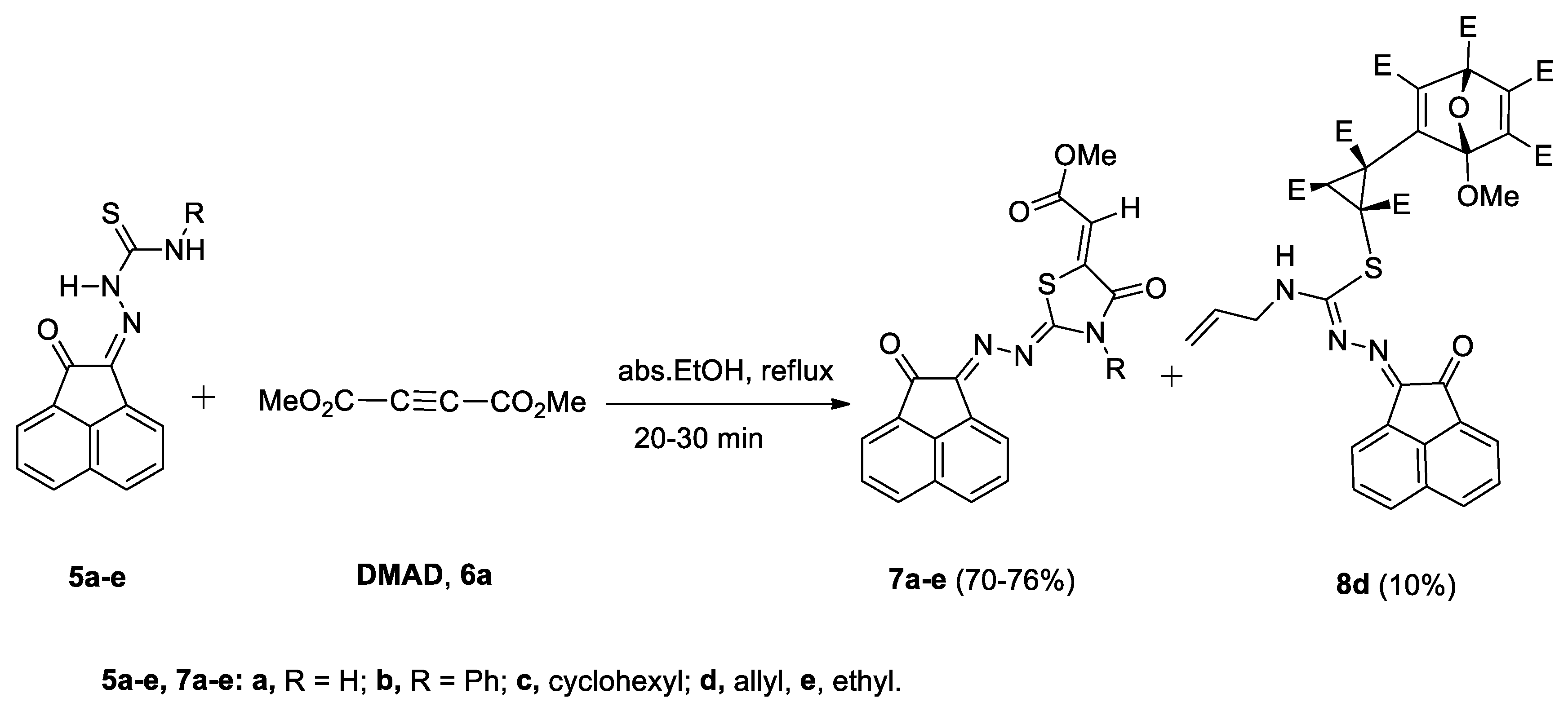
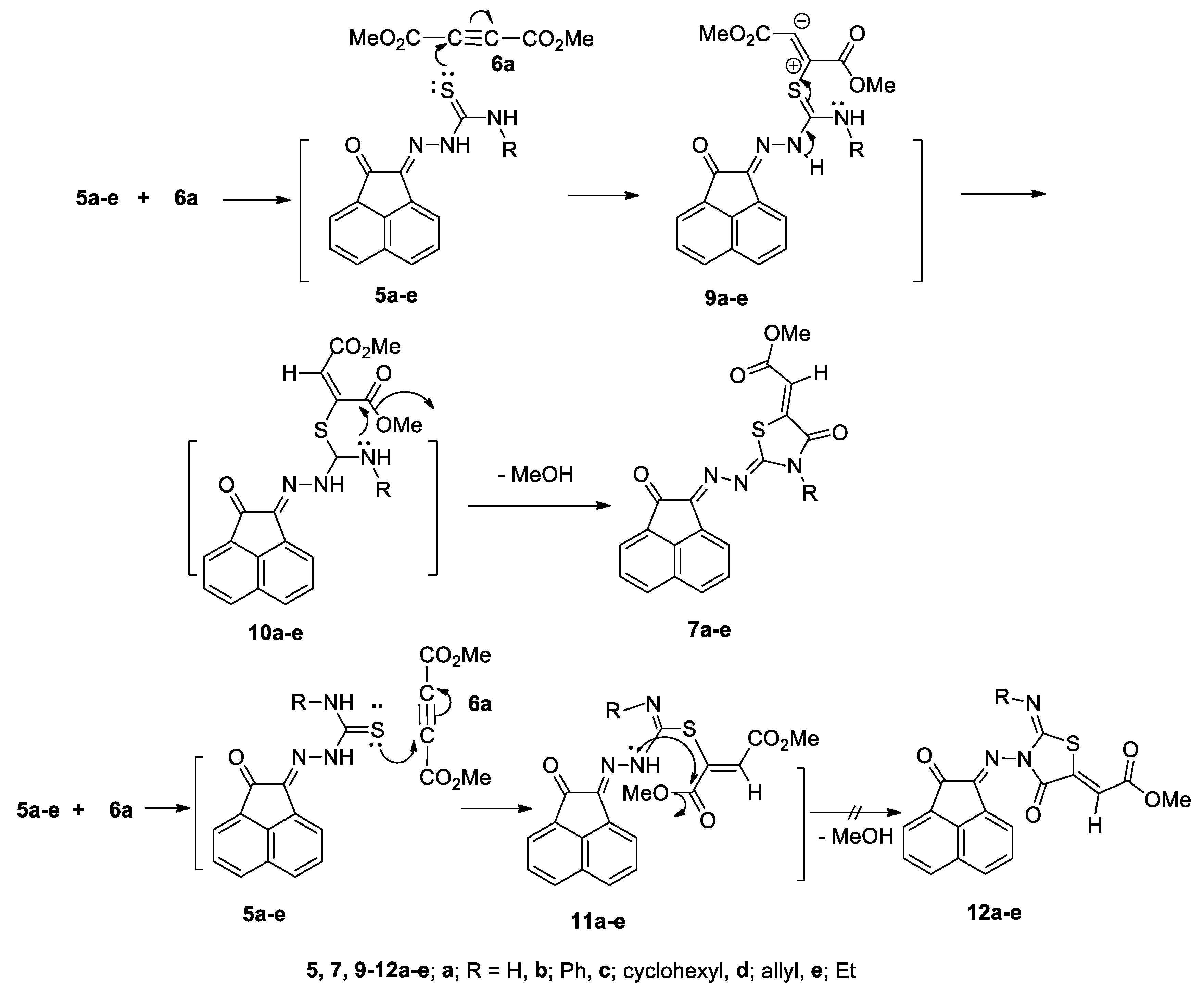
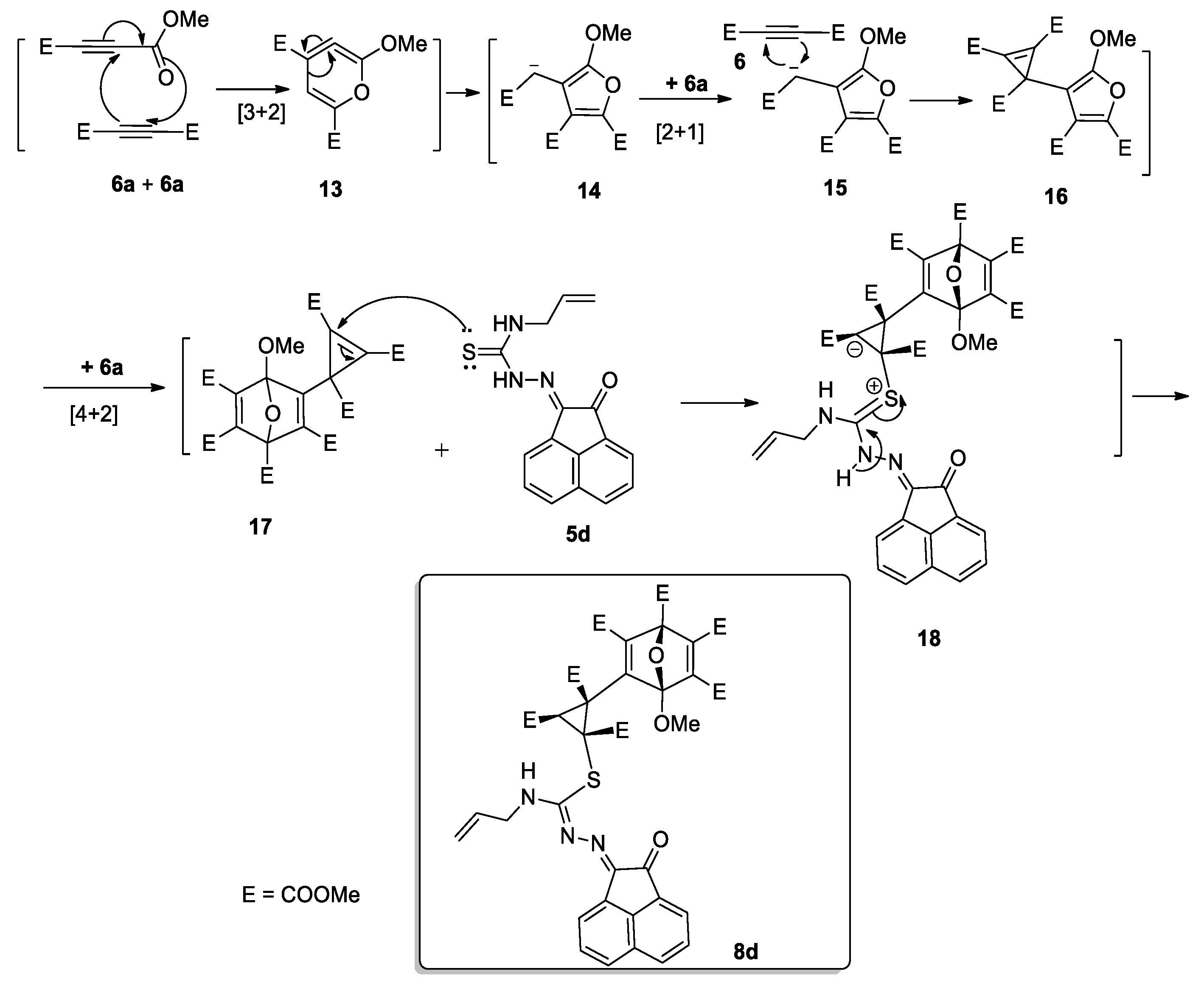
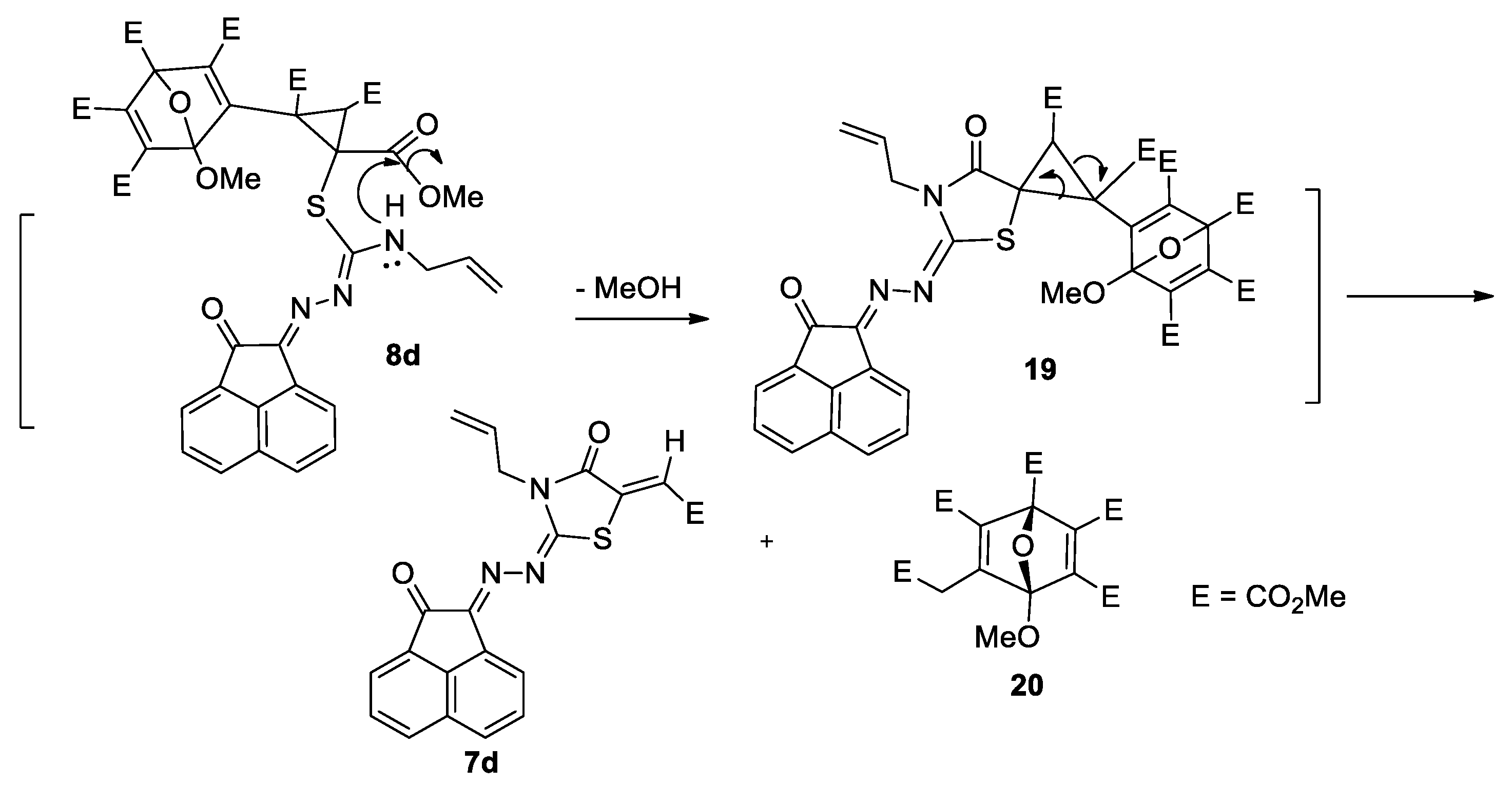
2.2. Reaction of Compounds 5a–e with Dimethyl Acetylenedicarboxylate (DMAD, 6)
3. Material and Methods
3.1. Chemistry
3.1.1. Starting Materials: Acenaphthequinone, 1 Was Bought and Bought from Aldrich, whereas [2.2]Paracyclophane Was Commercially Available
Preparation of 2-Oxoacenaphthylidene Thiosemicarbazones 5a–e
Reactions of 2-Oxoacenaphthylidene Thiosemicarbazone Derivatives with 6a
Preparation of 4-Acetyl-[2.2]Paracyclophanylidene-Thiosemicarbazones 22a,f
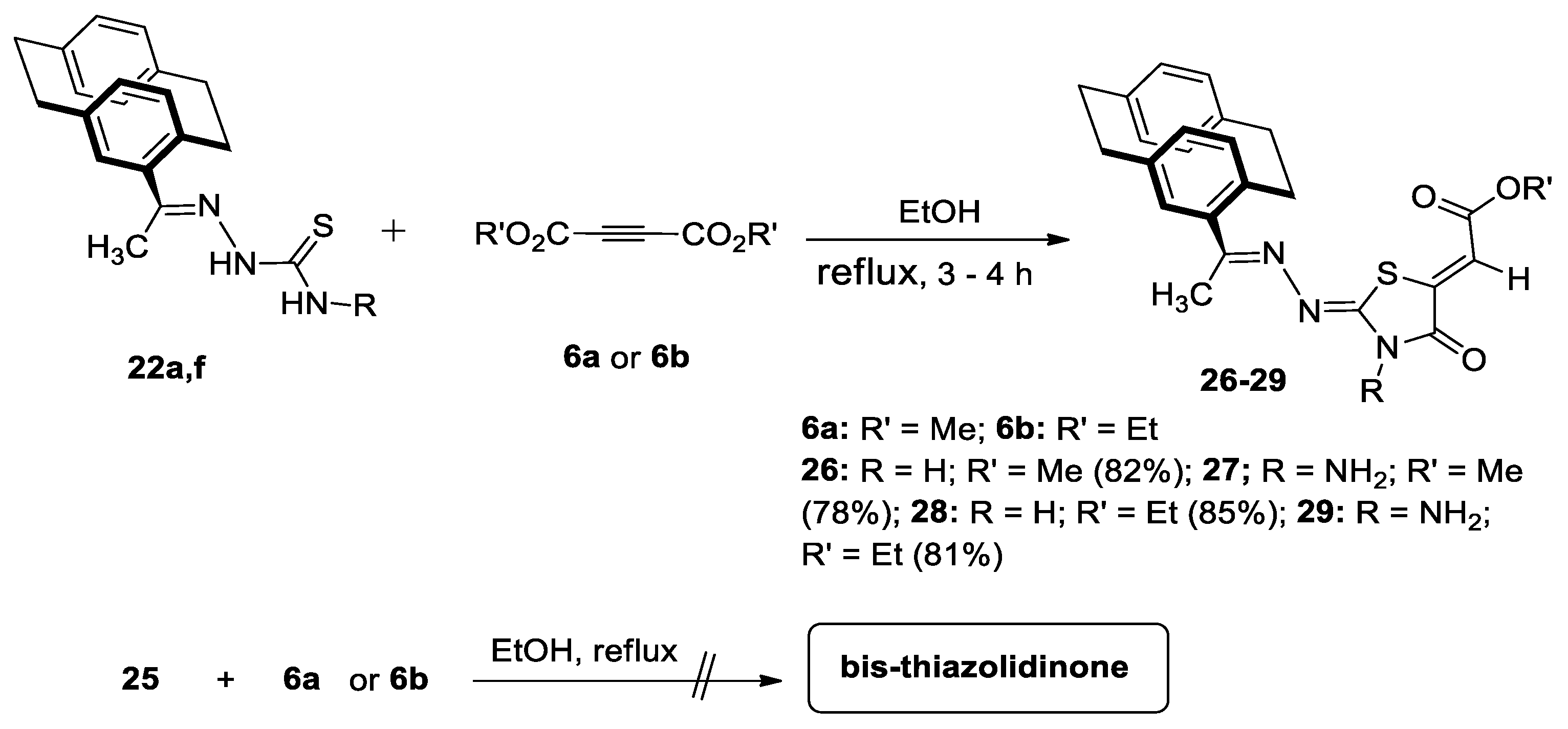
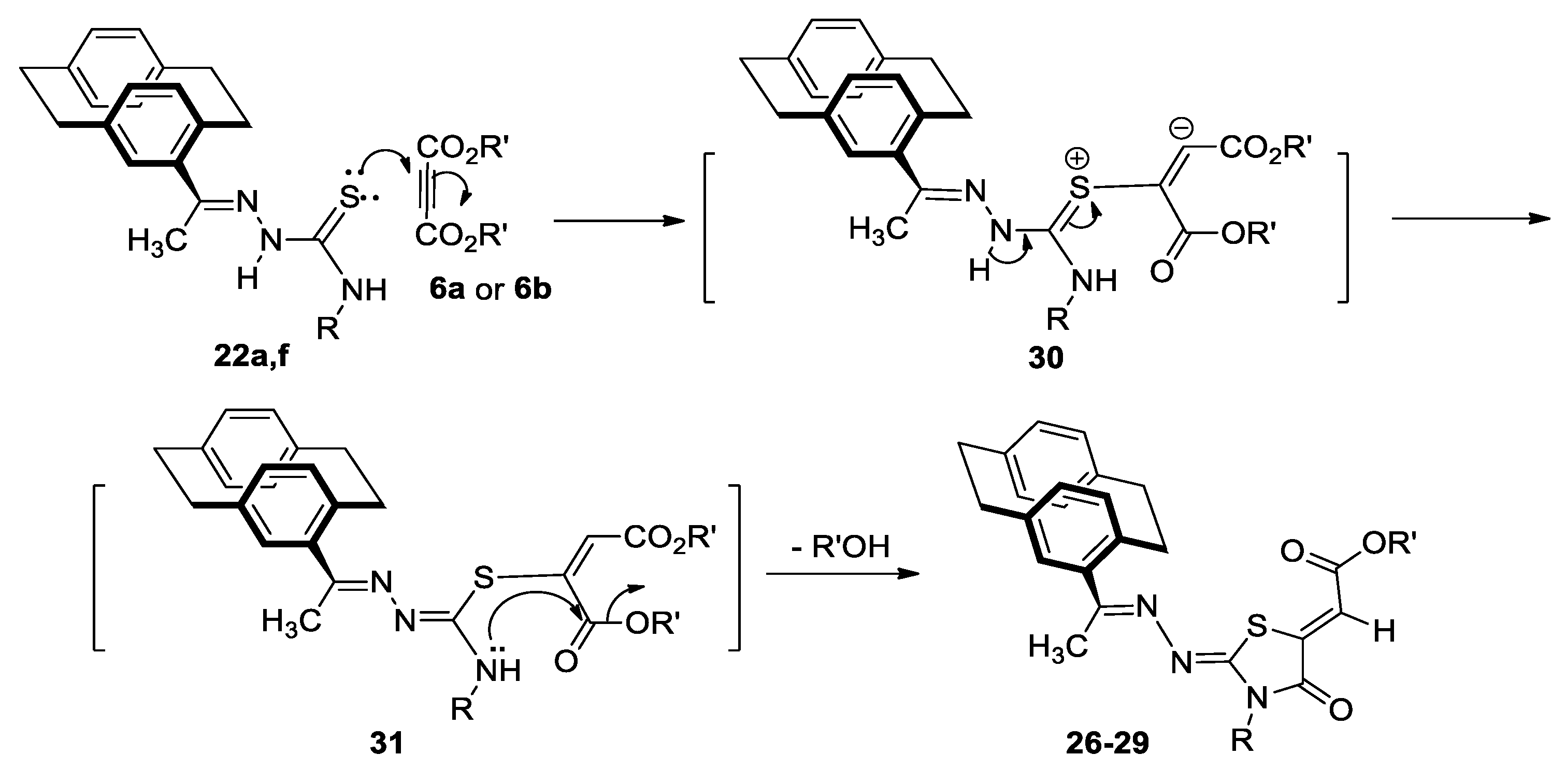
Reactions of 4-Acetyl[2.2]Paracyclophanylidene-Thiosemicarbazones, 22a,f with 6a and 6b
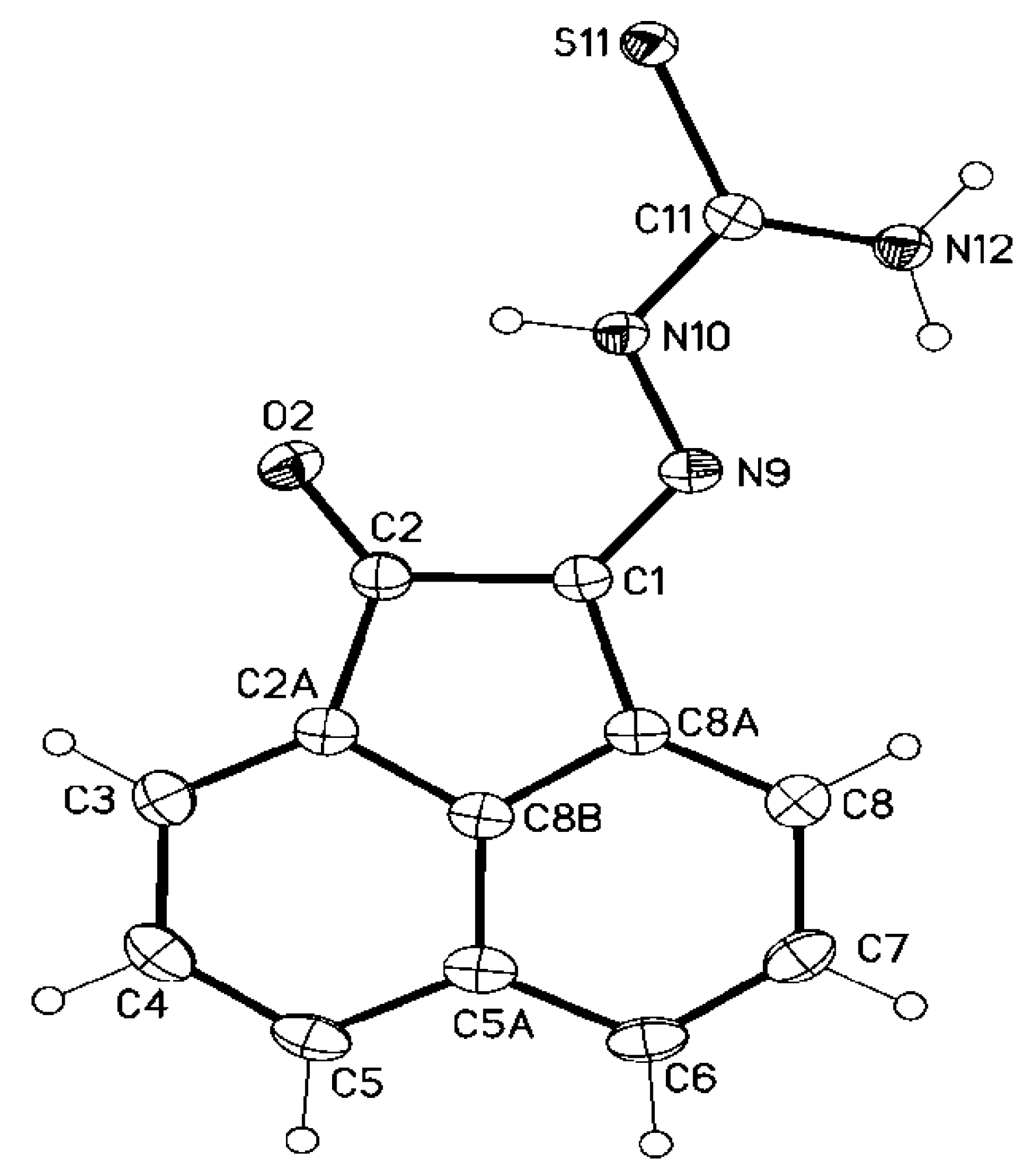
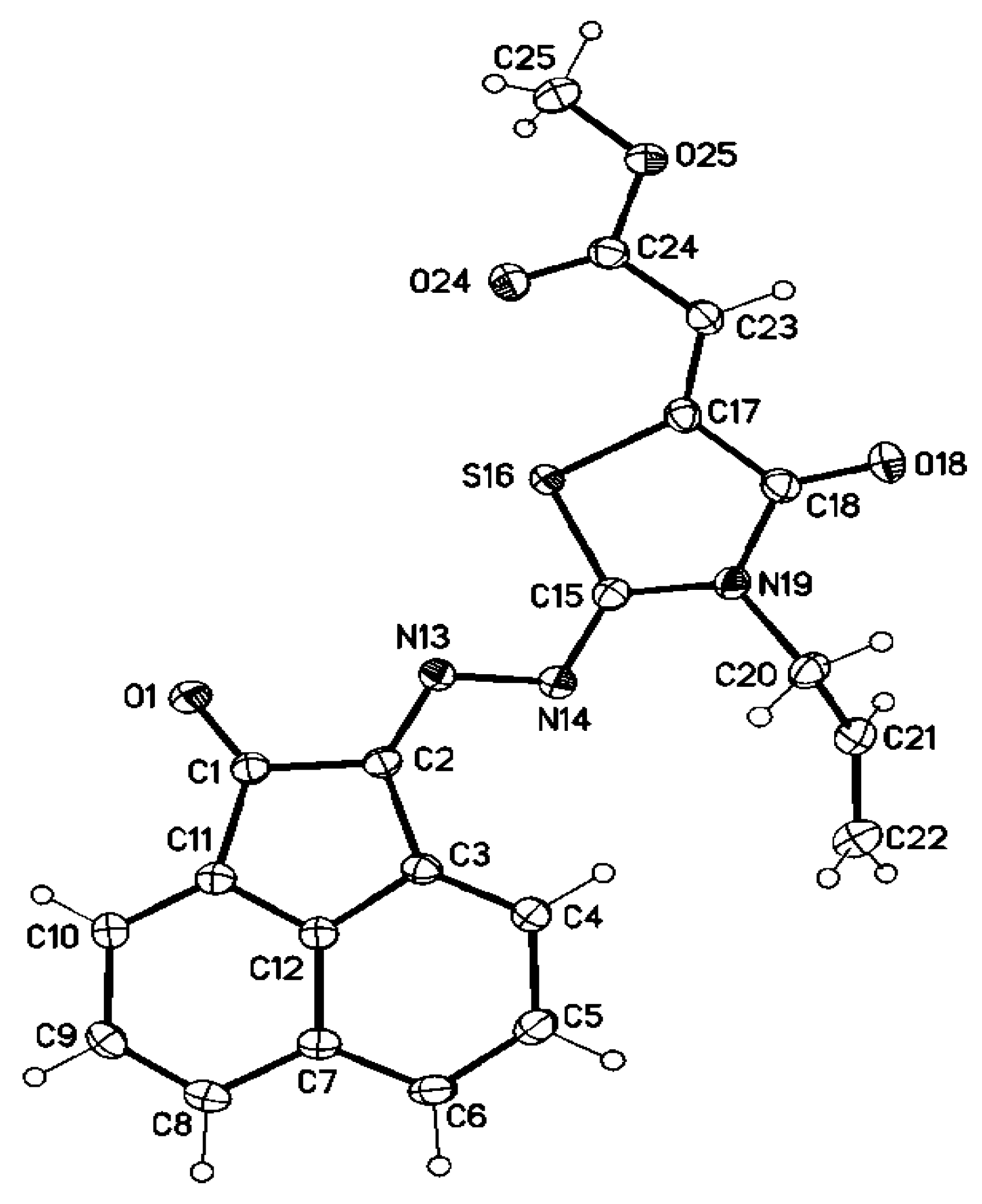
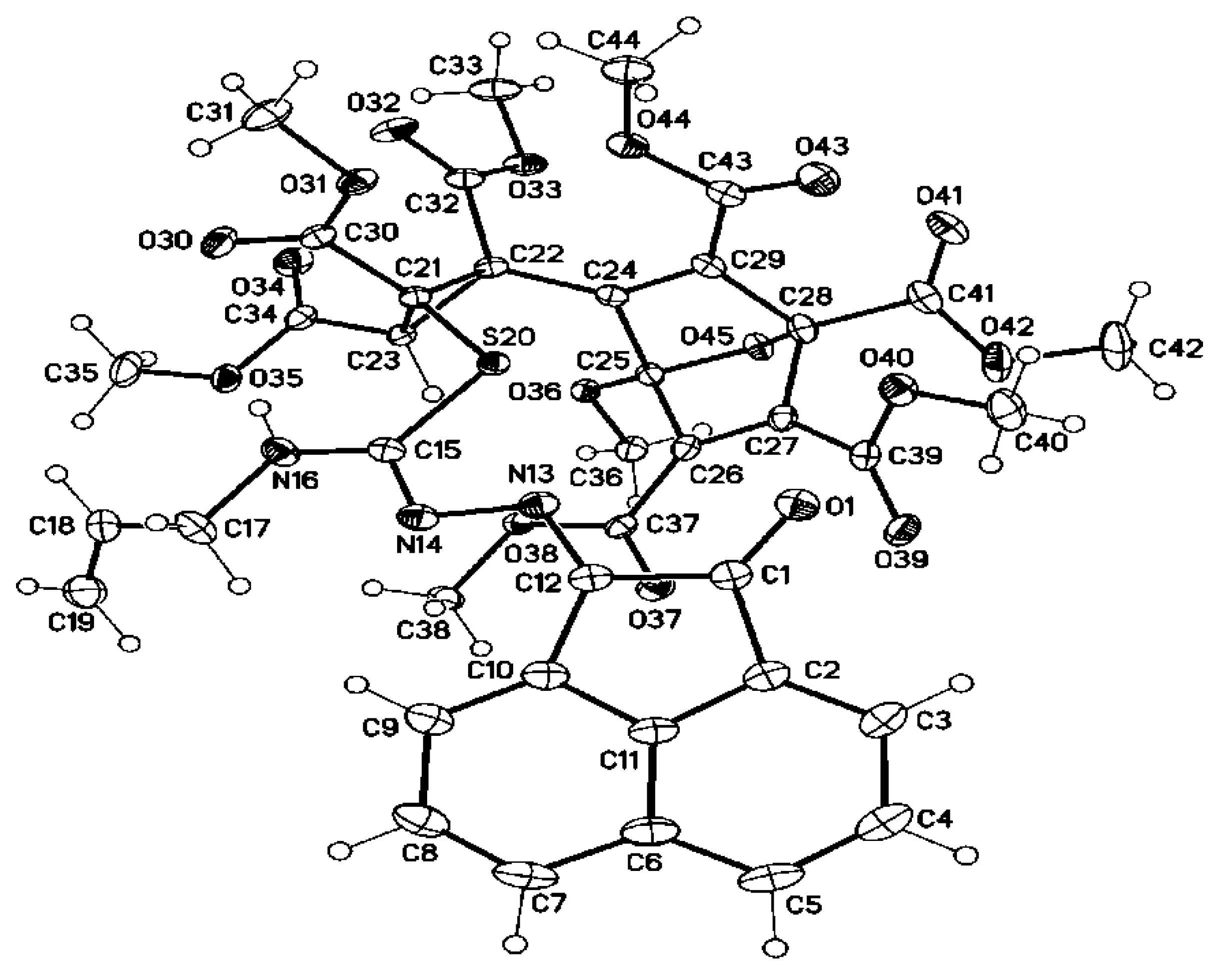
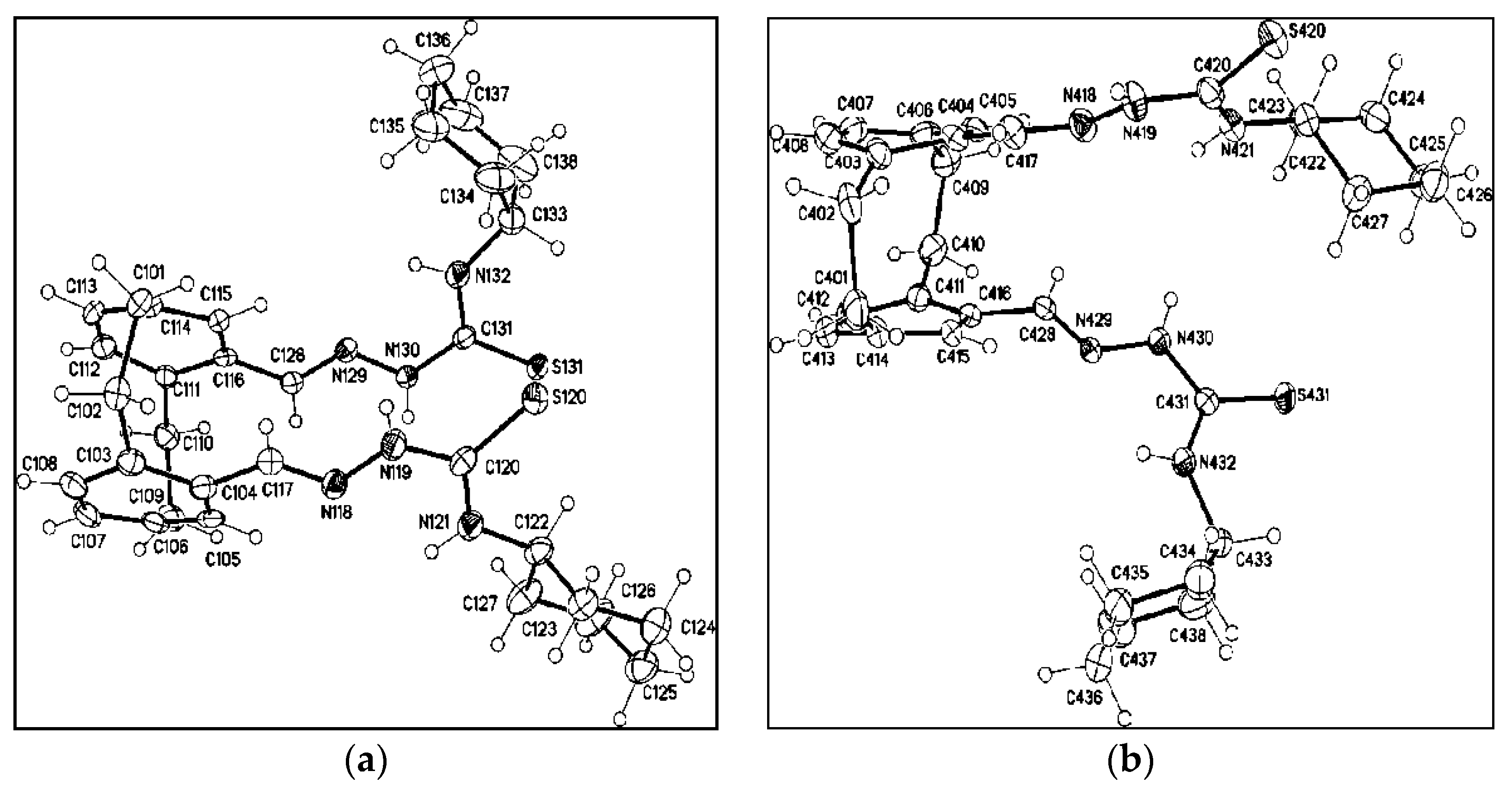
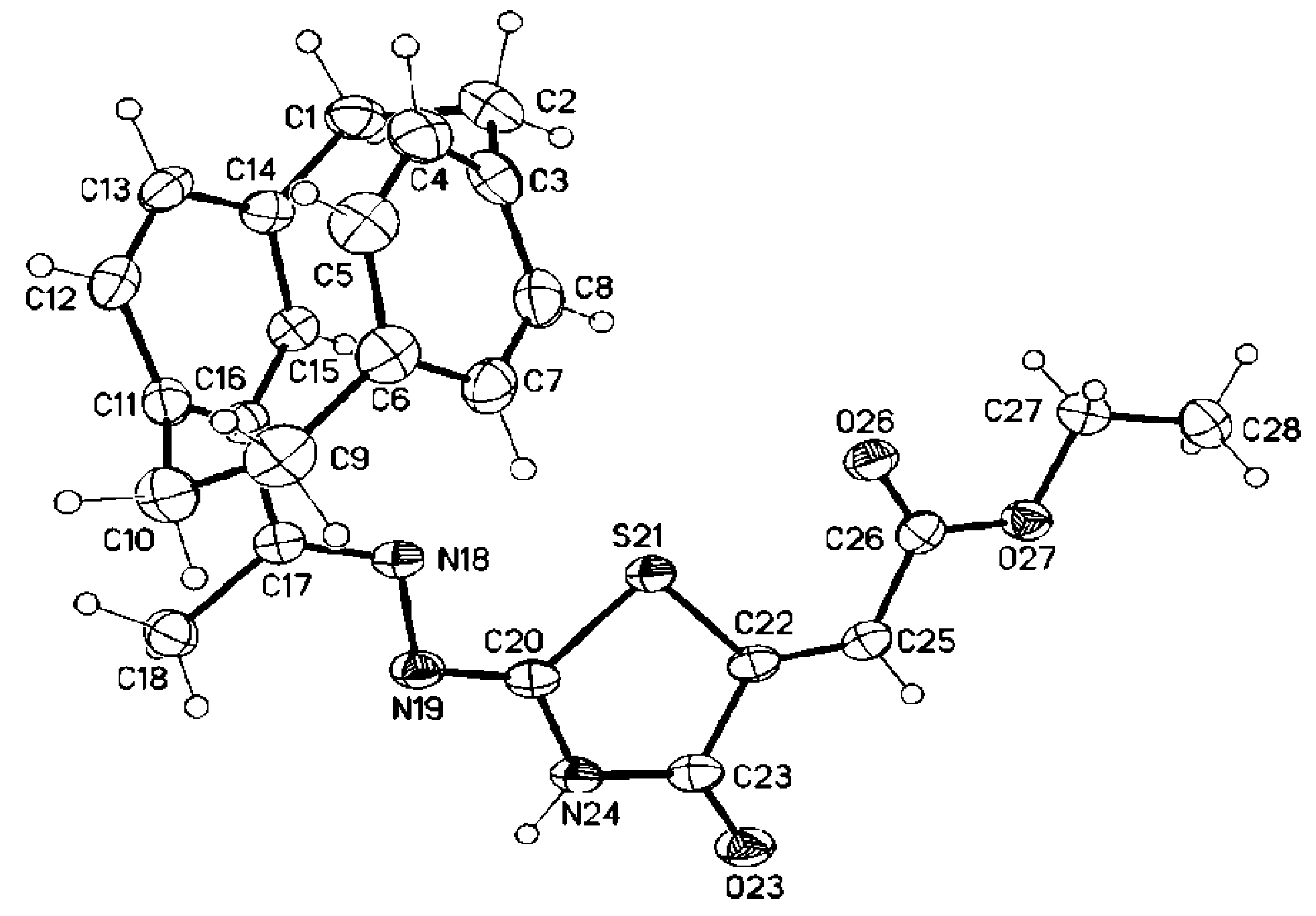
3.2. Single Crystal X-ray Structure Determination of 5a, 7d, 8d, 25 and 28
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Suthar, S.K.; Jaiswal, V.; Lohan, S.; Bansal, S.; Chaudhary, A.; Tiwari, A.; Alex, A.T.; Joseph, A. Novel quinolone substituted thiazolidin-4-ones as anti-inflammatory, anticancer agents: Design, synthesis and biological screening. Eur. J. Med. Chem. 2013, 63, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Aly, A.A.; Ishak, E.A.; El Malah, T.; Brown, A.B.; Elayat, W.M. Synthesis of potentially antioxidant and antibacterial biologically active thiazolidines. J. Heterocycl. Chem. 2015, 52, 1758–1764. [Google Scholar] [CrossRef]
- Sharma, S.; Sharma, P.K.; Kumar, N.; Dudhe, R. A review on various heterocyclic moieties and their antitubercular activity. Biomed. Pharmacother. 2011, 65, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Kumari, P.; Patel, N. Synthesis and biological evaluation of some thiazolidinones as antimicrobial agents. Eur. J. Med. Chem. 2012, 48, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Rawal, R.K.; Tripathi, R.; Katti, S.B.; Pannecouque, C.; de Clercq, E. Design, synthesis, and evaluation of 2-aryl-3-heteroaryl-1,3-thiazolidin-4-ones as anti-HIV agents. Bioorg. Med. Chem. 2007, 15, 1725–1731. [Google Scholar] [CrossRef] [PubMed]
- Shingalapur, R.V.; Hosamani, K.M.; Keri, R.S.; Hugar, M.H. Derivatives of benzimidazole pharmacophore: Synthesis, anticonvulsant, antidiabetic and DNA cleavage studies. Eur. J. Med. Chem. 2010, 45, 1753–1759. [Google Scholar] [CrossRef] [PubMed]
- Kishore, A.; Nampurath, G.K.; Mathew, S.P.; Zachariah, R.T.; Potu, B.K.; Rao, M.S.; Valiathan, M.; Chamallamudi, M.R. Antidiabetic effect through islet cell protection in streptozotocin diabetes: A preliminary assessment of two thiazolidin-4-ones in Swiss albino mice. Chem. Biol. Interact. 2009, 177, 242–246. [Google Scholar] [CrossRef]
- Aly, A.A.; Brown, A.B.; El-Emary, T.I.; Ewas, A.M.M.; Ramadan, M. Hydrazinecarbothioamide group in the synthesis of heterocycles. Arkivoc 2009, 2009, 150–197. [Google Scholar] [Green Version]
- Hassan, A.A.; Mohamed, N.K.; Makhlouf, M.M.; Bräse, S.; Nieger, M. Reactions of dimethyl acetylenedicarboxylate with 2,5-dithiobiurea derivatives. Synthesis 2014, 46, 3097–3102. [Google Scholar] [CrossRef]
- Hassan, A.A.; Mohamed, N.K.; Makhlouf, M.M.; Bräse, S.; Nieger, M.; Höpf, H. (Hex-2-en-ylidene)-N-substituted hydrazonecarbothioamides and 2,3-dichloro-1,4-naphthoquinone: Nucleophilic substitution reactions and synthesis of naphtho[2,3-f][1,3,4]-triazepines and naphtho[2,3-d]thiazoles. Synthesis 2016, 48, 3134–3140. [Google Scholar] [CrossRef]
- Aly, A.A.; Brown, A.B.; Abdel-Aziz, M.; Abuo-Rahma, G.E.D.A.; Radwan, M.F.; Ramadan, M.; Gamal-Eldeen, A.M. Synthesis of new 4-oxo-thiazolidine-5-ylidenes of antitumor and antioxidant activities. J. Heterocycl. Chem. 2010, 47, 547–554. [Google Scholar] [CrossRef]
- Aly, A.A.; Brown, A.B.; Abdel-Aziz, M.; Abuo-Rahma, G.E.D.A.; Radwan, M.F.; Ramadan, M.; Gamal-Eldeen, A.M. An efficient synthesis of thiazolidin-4-ones with antitumor and antioxidant activities. J. Heterocycl. Chem. 2012, 49, 726–731. [Google Scholar] [CrossRef]
- Hassan, A.A.; Abdel-Latif, F.F.; Nour El-Din, A.M.; Mostafa, S.M.; Nieger, M.; Bräse, S. Synthesis of (E)-2,5-disubstituted 1,3,4-thiadiazolyl-2,3-diphenylpropenones from alkenylidene-hydrazinecarbothioamides. Tetrahedron 2012, 68, 8487–8492. [Google Scholar] [CrossRef]
- Hu, W.-X.; Zhou, W.; Xia, C.-N.; Wen, X. Synthesis and anticancer activity of thiosemicarbazones. Bioorg. Med. Chem. Lett. 2006, 16, 2213–2218. [Google Scholar] [CrossRef] [PubMed]
- Barbuceanu, S.-F.; Ilies, D.C.; Saramet, G.; Uivarosi, V.; Draghici, C.; Radulescu, V. Synthesis and antioxidant activity evaluation of new compounds from hydrazinecarbothioamide and 1,2,4-triazole class containing diarylsulfone and 2,4-difluorophenyl moieties. Int. J. Mol. Sci. 2014, 15, 10908–10925. [Google Scholar] [CrossRef] [PubMed]
- Paiva, R.O.; Kneipp, L.F.; Goular, C.M.; Albuquerque, M.A.; Echevarria, A. Antifungal activities of thiosemicarbazones and semicarbazones against mycotoxigenic fungi. Ciência Agrotecnol. 2014, 38, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Reis, D.C.; Despaigne, A.A.R.; Da Silva, J.G.; Silva, N.F.; Vilela, C.F.; Mendes, I.C.; Takahashi, J.A.; Beraldo, H. Structural studies and investigation on the activity of imidazole-derived thiosemicarbazones and hydrazones against crop-related fungi. Molecules 2013, 18, 12645–12662. [Google Scholar] [CrossRef]
- El Ashry, E.S.H.; Abdel Hamid, H.; Kassem, A.A.; Shoukry, M. Synthesis and reactions of acenaphthenequinones. Part 2. The reactions of acenaphthenequinones. Molecules 2002, 7, 155–188. [Google Scholar] [CrossRef]
- Mhaidat, I.; Mergos, J.A.; Hamilakis, S.; Kollia, C.; Loizos, Z.; Tsolomitis, A.; Dervos, C.T. Synthesis and reactions of acenaphthenequinones. Part 2. The reactions of acenaphthenequinones. Mater. Lett. 2009, 63, 2587–2590. [Google Scholar] [CrossRef]
- Ziarani, G.M.; Hajiabbasi, P.; Gholamzadeh, P. Development of the acenaphthenequinone reactions. Heterocycles 2012, 85, 1869–1890. [Google Scholar] [CrossRef]
- Yavari, I.; Khajeh-Khezri, A. Recent advances in the synthesis of hetero- and carbocyclic compounds and complexes based on acenaphthylene-1,2-dione. Synthesis 2018, 50, 3947–3973. [Google Scholar] [CrossRef]
- Hyatt, J.L.; Wadkins, R.M.; Tsurkan, L.; Hicks, L.D.; Hatfield, M.J.; Edwards, C.C.; Ross, C.R., II; Cantalupo, S.A.; Crundwell, G.; Danks, M.K.; et al. Planarity and constrain of the carbonyl groups in 1,2-diones are determinants for selective inhibition of human carboxylesterase 1. J. Med. Chem. 2007, 50, 5727–5734. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Argüelles, M.C.; Ferrari, M.B.; Fava, G.G.; Pelizzi, C.; Pelosi, G.; Albertini, R.; Bonati, A.; Dall’Aglio, P.P.; Lunghi, P.; Pinelli, S. Acenaphthenequinone thiosemicarbazone and its transition metal complexes: Synthesis, structure, and biological activity. J. Inorg. Biochem. 1997, 66, 7–17. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, H.; Wu, G.; Li, Z.; Song, T.; Li, X.Q. Probing the difference between BH3 groove of Mcl-1 and Bcl-2 protein: Implications for dual inhibitors design. Eur. J. Med. Chem. 2011, 46, 3909–3916. [Google Scholar] [CrossRef] [PubMed]
- El-Alawi, Y.S.; McConkey, B.J.; Dixon, D.G.; Greenberg, B.M. Measurement of short- and long-term toxicity of polycyclic aromatic hydrocarbons using luminescent bacteria. Ecotoxicol. Environ. Saf. 2002, 51, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.; Biju, A.T.; Vinod, A.U.; Suresh, E. Reaction of Huisgen Zwitter ion with 1,2-benzoquinones and isatins: Synthesis of dihydro-1,2,3-benzoxadiazoles and spirooxadiazolines. Org. Lett. 2005, 7, 5139–5142. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jiang, Y.-H.; Yan, C-G. Convenient synthesis of spiro (indoline-3,4′-pyrano[2,3-c]pyrazole) and spiro (acenaphthyl-3,4′-pyrano[2,3-c]pyrazoles via four-component reaction. Chin. Chem. Lett. 2015, 26, 889–893. [Google Scholar] [CrossRef]
- Yavari, I.; Baoosi, L.; Halvagar, M.R. A synthesis of fused acenaphthopyrrolizines via the 1,3-dipolar cycloaddition reaction of azomethine ylides with acetylenic esters. Mol. Divers. 2017, 21, 257–263. [Google Scholar] [CrossRef]
- Wei, A.C.; Ali, M.A.; Yoon, Y.K.; Ismail, R.; Choon, T.S.; Kumar, R.S. A facile three-component [3+2]-cycloaddition for the regioselective synthesis of highly functionalized dispiropyrrolidines acting as antimycobacterial agents. Bioorg. Med. Chem. Lett. 2013, 23, 1383–1386. [Google Scholar] [CrossRef]
- Arumugam, N.; Almansour, A.I.; Kumar, R.S.; Perumal, S.; Ghabbour, H.A.; Fun, H.-K. A 1,3-dippolar cycloaddition-annulation protocol for the expedient region-, stereo-, and product-selective construction of novel hybrid heterocycles comprising seven rings and seven contiguous stereocentres. Tetrahedron Lett. 2013, 54, 2515–2519. [Google Scholar] [CrossRef]
- Song, L.-L.; Yang, C.; Yu, Y.-Q.; Xu, D.-Z. A simple and green tandem Knoevenagel-phospha-Michael reaction for one-pot synthesis of 2-oxindol-3-yl-phosphonates catalyzed by a DABCO-based ionic liquid. Synthesis 2017, 49, 1641–1647. [Google Scholar]
- Yavari, I.; Baoosi, L.; Halvagar, M.R. A convenient synthesis of fused tetrahydroazocines from acenaphthylene-1,2-dione, proline, and acetylenic esters. Synlett 2018, 29, 635–639. [Google Scholar] [CrossRef]
- Wang, X.-H.; Yan, C.-G. Facile synthesis of spiro(indane-2,1′-pyrrolo[2,1-a]isoquinolines) via three-component reaction of isoquinolinium salts, indane-1,3-dione, and isatins. Synthesis 2014, 46, 1059–1066. [Google Scholar] [CrossRef]
- Gong, H.; Sun, J.; Yan, C.-G. Efficient synthesis of polycyclic dispirooxindoles via domino Diels-Alder cyclodimerization reaction. Tetrahedron 2014, 70, 6641–6650. [Google Scholar] [CrossRef]
- Ahadi, S.; Hosseini, G.; Bazgir, A. Synthesis of oxo-indolin-3-ylidene-1,3-dithioles. J. Iran. Chem. Soc. 2012, 9, 333–338. [Google Scholar] [CrossRef]
- Kiruthika, S.E.; Lakshmi, N.V.; Banu, B.R.; Perumal, P.T. A facile strategy for the one pot multicomponent synthesis of spiro dihydropyridines from amines and activated alkynes. Tetrahedron Lett. 2011, 52, 6508–6511. [Google Scholar] [CrossRef]
- Hopf, H. [2.2] Paracyclophanes in polymer chemistry and materials science. Angew. Chem. Int. Ed. 2008, 47, 9808–9812. [Google Scholar] [CrossRef] [PubMed]
- Gibson, S.E.; Knight, J.D. [2.2] Paracyclophane derivatives in asymmetric catalysis. Org. Biomol. Chem. 2003, 1, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Gulder, T.; Baran, P.S. Strained cyclophane natural products: Macrocyclization at its limits. Nat. Prod. Rep. 2012, 29, 899–934. [Google Scholar] [CrossRef] [PubMed]
- Hassan, Z.; Spuling, E.; Knoll, D.M.; Bräse, S. Regioselective functionalization of [2.2] paracyclophanes: Recent synthetic progress and perspectives. Angew. Chem. Int. Ed. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Aly, A.A.; Brown, A.B. Asymmetric and fused heterocycles based on [2.2] paracyclophane. Tetrahedron 2009, 65, 8055–8089. [Google Scholar] [CrossRef]
- Aly, A.A.; Hopf, H.; Ernst, L.; Dix, I.; Jones, P.G. New cycloadditions of (E)-N,α-dimethyl-α-(4-[2.2] paracyclophanyl)nitrone. Eur. J. Org. Chem. 2006, 2006, 3001–3006. [Google Scholar] [CrossRef]
- Aly, A.A. Cycloaddition of (E)-N-{2-([2.2]paracyclophan-4-yl)ethylidene}methylamine N-oxide with 2,3-diphenylcyclopropenones and dibenzoyl acetylene: Synthesis of new paracyclophanylpyrroles. J. Chem. Res. 2007, 2007, 451–454. [Google Scholar] [CrossRef]
- Aly, A.A.; Hopf, H.; Jones, P.G.; Dix, I. Cycloadditions of α-(4-[2.2]paracyclophane)-N-methyl nitrone. Tetrahedron 2006, 62, 4498–4505. [Google Scholar] [CrossRef]
- Hopf, H.; Aly, A.A.; Swaminathan, V.N.; Ernst, L.; Dix, I.; Jones, P.G. A simple route to a pyridinyl[2.2]paracyclophane. Eur. J. Org. Chem. 2005, 2005, 68–71. [Google Scholar] [CrossRef]
- Aly, A.A.; Bräse, S.; Weis, P. Tridentate and bidentate copper complexes of [2.2] paracyclophanyl-substituted thiosemicarbazones, thiocarbazones, hydrazones, and thioureas. J. Mol. Struct. 2019, 1178, 311–326. [Google Scholar] [CrossRef]
- Pascu, S.L.; Waghorn, P.A.; Churchill, G.C.; Sim, R.B. Synthesis of Metal Complexes with Thiosemicarbazone Derivatives for Use in Medical Imaging and Therapy. PCT International Application WO 2008025941 A2 20080306, 4 August 2008. [Google Scholar]
- Patil, P.B.; Patil, J.D.; Korade, S.N.; Kshirsagar, S.D.; Govindwar, S.P.; Pore, D.M. An efficient synthesis of anti-microbial 1,2,4-triazole-3-thiones promoted by acidic ionic liquid. Res. Chem. Intermed. 2016, 42, 4171–4180. [Google Scholar] [CrossRef]
- Mohammadi, M.K.; Firuzi, O.; Khoshneviszadeh, M.; Razzaghi-Asl, N.; Sepehri, S.; Miri, R. Novel 9-(alkylthio)-acenaphtho[1,2-e]-1,2,4-triazine derivatives: Synthesis, cytotoxic activity, and molecular docking studies on B-cell lymphoma 2 (Bcl-2). DARU J. Pharm. Sci. 2014, 22, 2. [Google Scholar] [CrossRef] [PubMed]
- Satheshkumar, A.; El-Mossalamy, E.H.; Manivannam, R.; Parthiban, C.; Al-Harbi, L.M.; Kosa, S.; Elango, K.P. Anion induced azo-hydrazine tautomerism for the selective colorimetric sensing of fluoride ion. Spectrochim. Acta A 2014, 128, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Kauer, J.C.; Simmons, H.E. Tetramers of acetylenedicarboxylic esters. J. Org. Chem. 1968, 33, 2720–2726. [Google Scholar] [CrossRef]
- Hocking, M.B.; van der Voort Maarschalk, F.W. X-ray structures of triphenylphosphine and 1,3,5-triphenylphosphole products with dimethyl acetylenedicarboxylate tetramer. Can. J. Chem. 1994, 72, 2428–2442. [Google Scholar] [CrossRef]
- Winterfeldt, E.; Giesler, G. Formation of trimethyl 2-methoxyfurantricarboxylate from dimethyl acetylenedicarboxylate. Angew. Chem. Int. Ed. 1966, 5, 579. [Google Scholar] [CrossRef]
- Banert, K.; Bochmann, S.; Ihle, A.; Plefka, O.; Taubert, F.; Walther, T.; Korb, M.; Rueffer, T.; Lang, H. Synthesis with perfect atom economy: Generation of furan derivatives by 1,3-dipolar cycloaddition of acetylenedicarboxylates at cyclooctynes. Molecules 2014, 19, 14022–14035. [Google Scholar] [CrossRef] [PubMed]
- Medina, S.; Gonzalez-Gomez, A.; Dominguez, G.; Perez-Castells, J. Medium-sized and strained heterocycles from non-catalyzed and gold-catalyzed conversions of β-carbolines. Org. Biomol. Chem. 2012, 10, 7167–7176. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Luo, H.; Tao, G.; Cai, W.; Cao, J.; Duan, Z.; Mathey, F. Selective synthesis of (Z)-diazadiphosphafulvalene from 2,2′-bis-azaphosphindole. Org. Lett. 2018, 20, 1027–1030. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, R.L.; Waghorn, P.A.; Jones, M.W.; Bauman, A.; Brayshaw, S.K.; Hu, Z.; Kociok-Köhn, G.; Mindt, T.L.; Tyrrell, R.M.; Botchway, S.W.; et al. Fluorescent gallium and indium bis(thiosemicarbazonates) and their radiolabeled analogues: Synthesis, structures, and cellular confocal imaging investigations. Dalton Trans. 2011, 40, 6238–6252. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Tadjarodi, A.; Najjari, S.; Notash, B. Synthesis and crystal structure of a new thiosemicarbazone, acenaphthenequinone thiosemiscarbazone mono methanol. Iran. J. Crystallogr. Miner. 2015, 22, 109–114. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
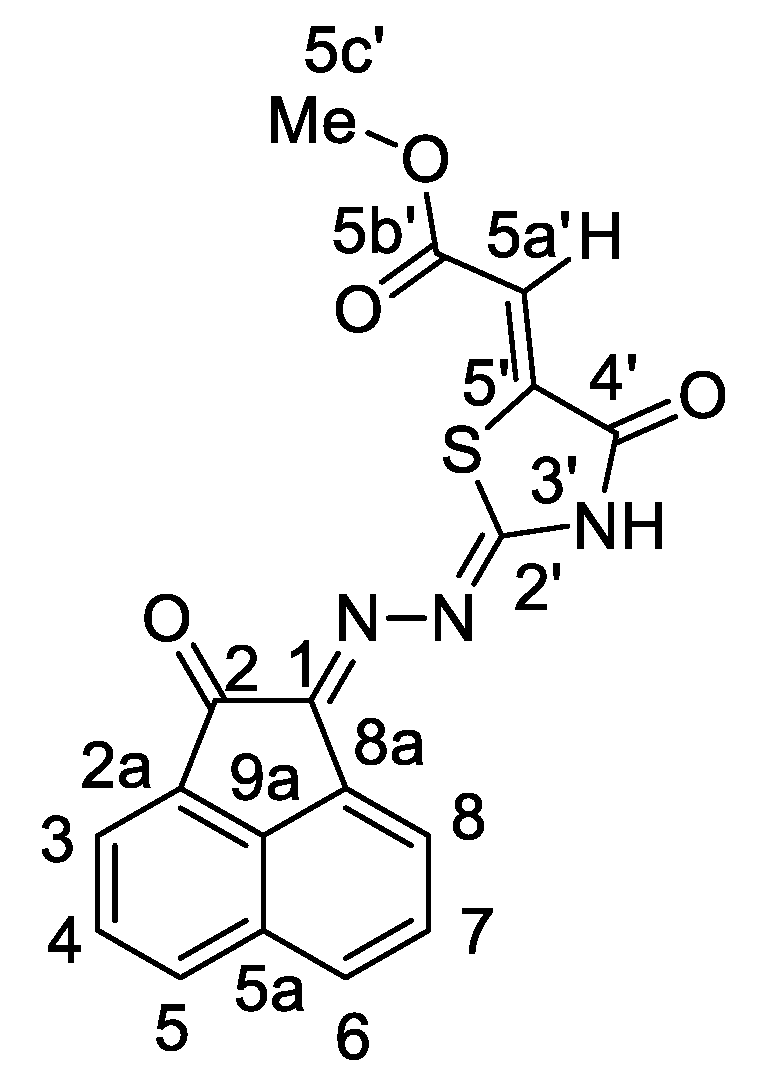
| 1H-NMR: | 1H-1H COSY: | Assignment: | |
| 13.46 (b; 1H) | NH-3′ | ||
| 8.35 (d, J = 8.2; 1H) | 7.86 | H-5 | |
| 8.20 (d, J = 8.2; 1H) | 7.85 | H-6 | |
| 8.01 (d, J = 7.8; 1H) | 7.86 | H-3 | |
| 7.99 (d, J = 7.0; 1H) | 7.85 | H-8 | |
| 7.86 (“t”, J = 7.9; 1H)8.35, 8.01 | H-4 | ||
| 7.85 (“t”, J = 8.0; 1H) | 8.20, 7.99 | H-7 | |
| 6.73 (s; 1H) | H-5a′ | ||
| 3.81 (s; 3H) | H-5c′ | ||
| 13C NMR: | 1H-13C HSQC: H-13C HMBC: | Assignment: | |
| 183.77 | 8.01, 7.86 | C-2 | |
| 166.01 | C-4 | ||
| 165.78 | 6.73, 3.81 | C-5′ | |
| 154.23 | C-2′ | ||
| 151.66 | 7.99 | C-1 | |
| 142.07 | 6.73 | C-5′ | |
| 139.71 | 8.35, 8.20, 8.01, 7.86, 7.85 | C-9a | |
| 131.86 | 8.35 | 8.20, 8.01, 7.86 | C-5 |
| 131.15, 131.05, 130.06 | 8.35, 8.20, 8.01, 7.86, 7.85 | C-2a, 5a, 9 | |
| 128.95, 128.59 | 7.86, 7.85 | 8.35, 7.99 | C-4, 7 |
| 128.20 | 8.20 | 8.20 | C-6 |
| 121.52 | 8.01 | 8.35 | C-3 |
| 118.82 | 7.99 | 8.20 | C-8 |
| 115.27 | 6.73 | 6.73, 3.81 | C-5a′ |
| 52.49 | 3.81 | 3.81 | C-5c′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aly, A.A.; Mohamed, N.K.; Hassan, A.A.; El-Shaieb, K.M.; Makhlouf, M.M.; Bräse, S.; Nieger, M.; Brown, A.B. Functionalized 1,3-Thiazolidin-4-Ones from 2-Oxo-Acenaphthoquinylidene- and [2.2]Paracyclophanylidene-Thiosemicarbazones. Molecules 2019, 24, 3069. https://doi.org/10.3390/molecules24173069
Aly AA, Mohamed NK, Hassan AA, El-Shaieb KM, Makhlouf MM, Bräse S, Nieger M, Brown AB. Functionalized 1,3-Thiazolidin-4-Ones from 2-Oxo-Acenaphthoquinylidene- and [2.2]Paracyclophanylidene-Thiosemicarbazones. Molecules. 2019; 24(17):3069. https://doi.org/10.3390/molecules24173069
Chicago/Turabian StyleAly, Ashraf A., Nasr K. Mohamed, Alaa A. Hassan, Kamal M. El-Shaieb, Maysa M. Makhlouf, Stefan Bräse, Martin Nieger, and Alan B. Brown. 2019. "Functionalized 1,3-Thiazolidin-4-Ones from 2-Oxo-Acenaphthoquinylidene- and [2.2]Paracyclophanylidene-Thiosemicarbazones" Molecules 24, no. 17: 3069. https://doi.org/10.3390/molecules24173069