
The Importance of Reaction Conditions on the Chemical Structure of N,O-Acylated Chitosan Derivatives



Abstract
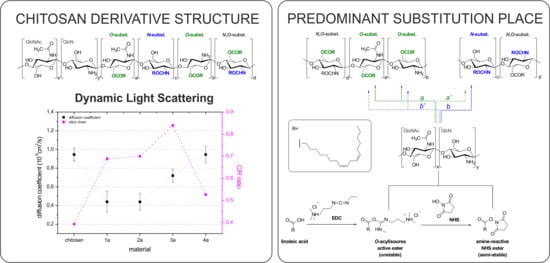
1. Introduction
2. Results and Discussion
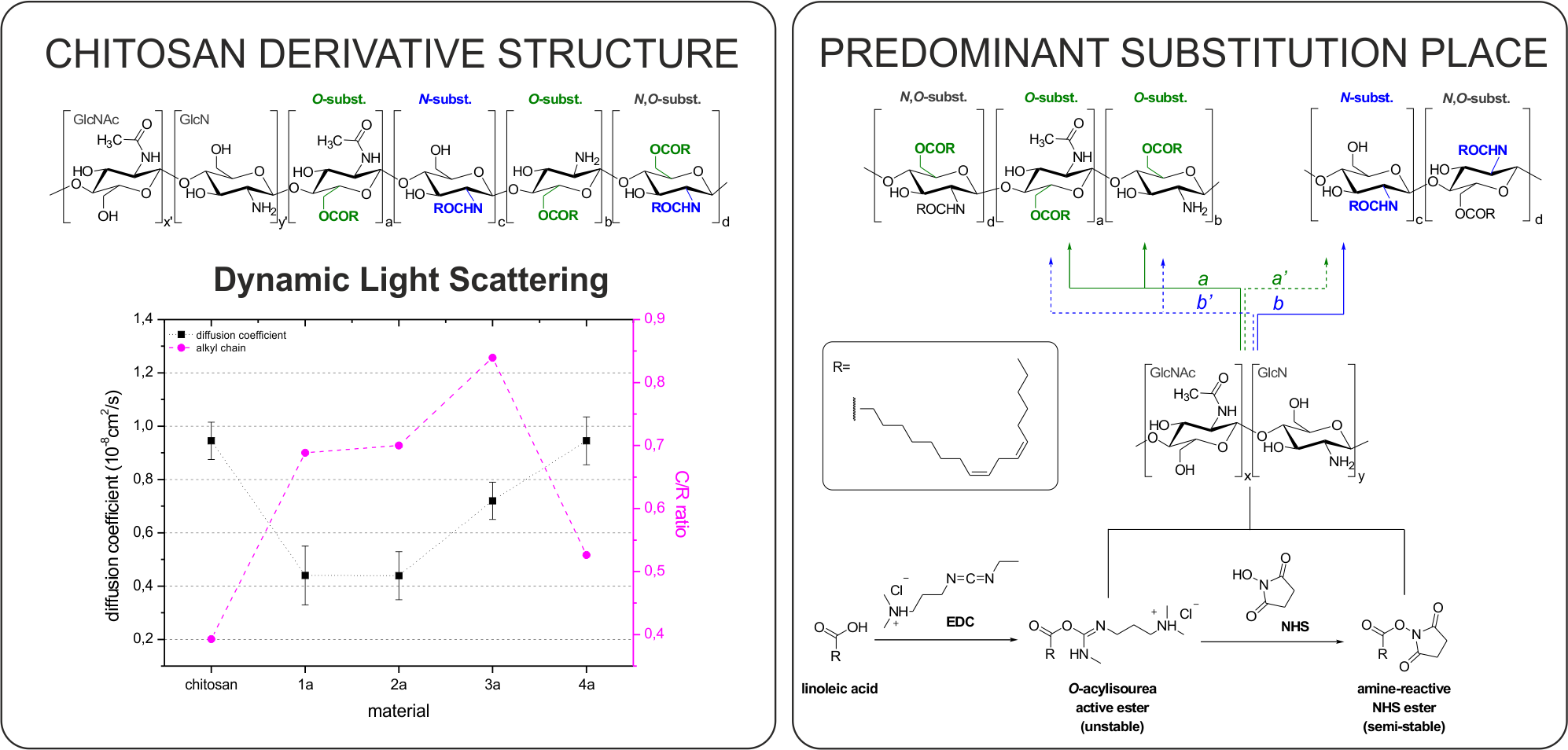
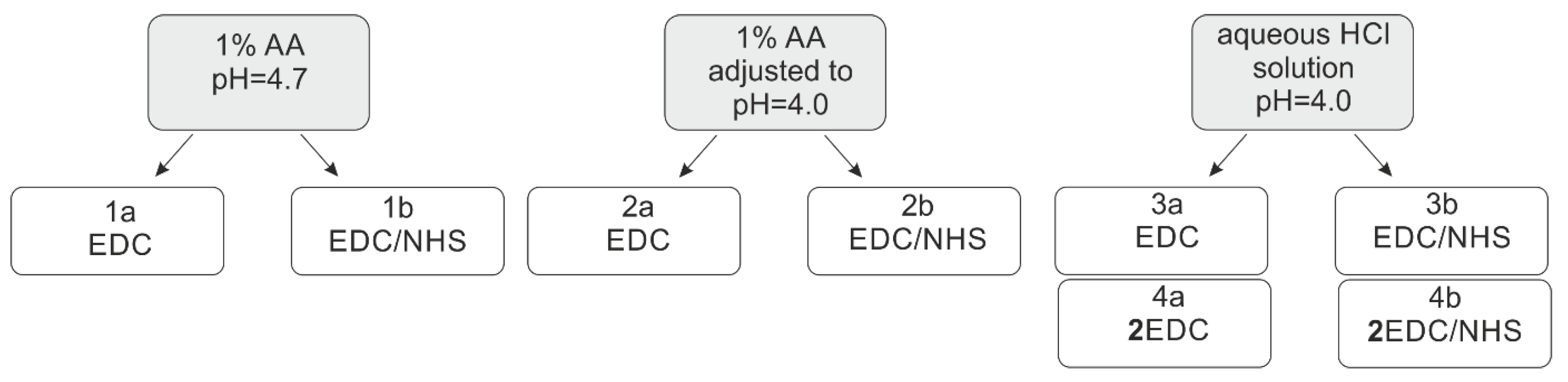
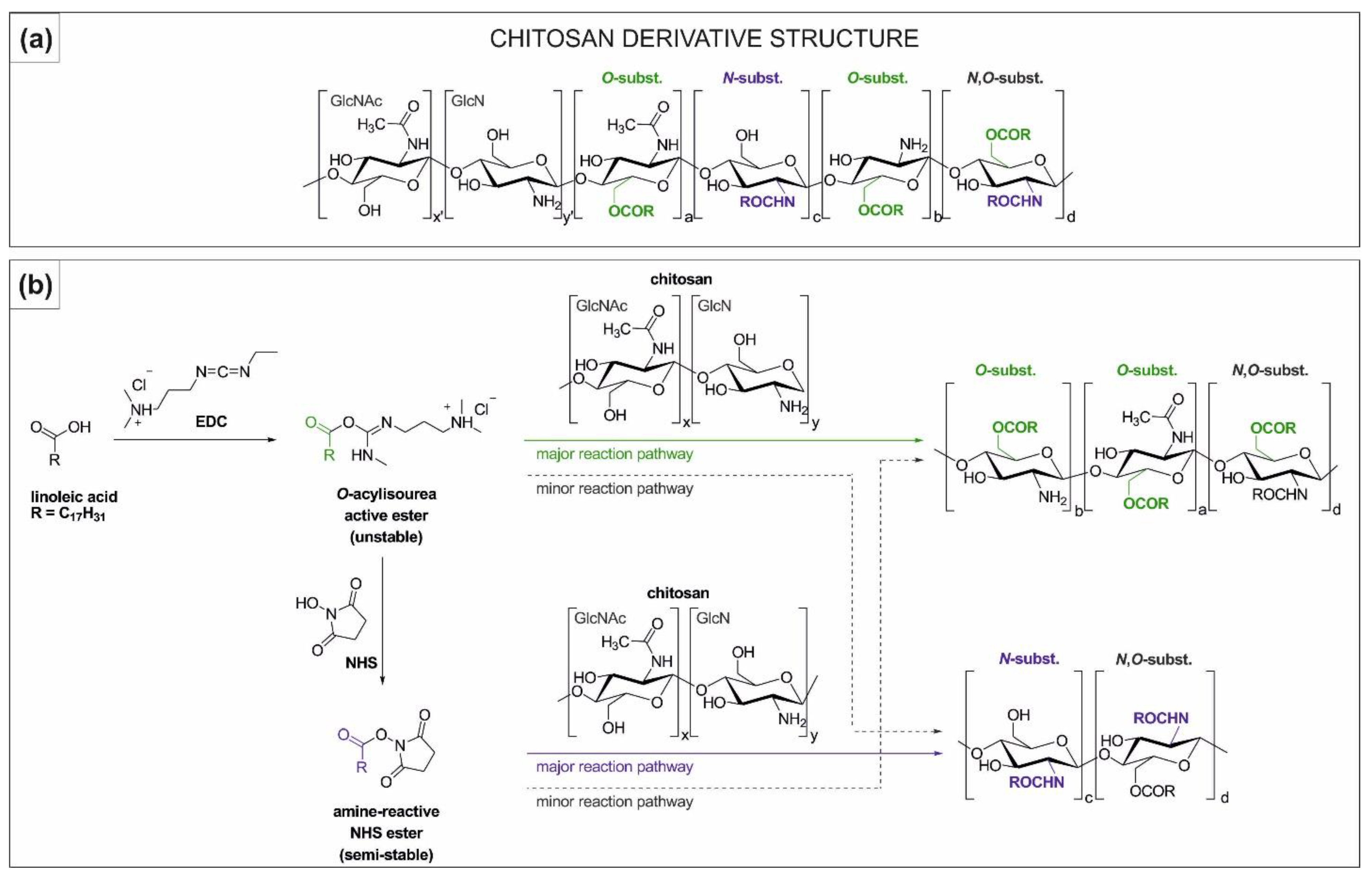
2.1. Synthesis and Characterisation of Chitosan Derivatives—Effect of Reaction Conditions over Acylation Type (N–, O–), Substitution Degree and Chemical Structure
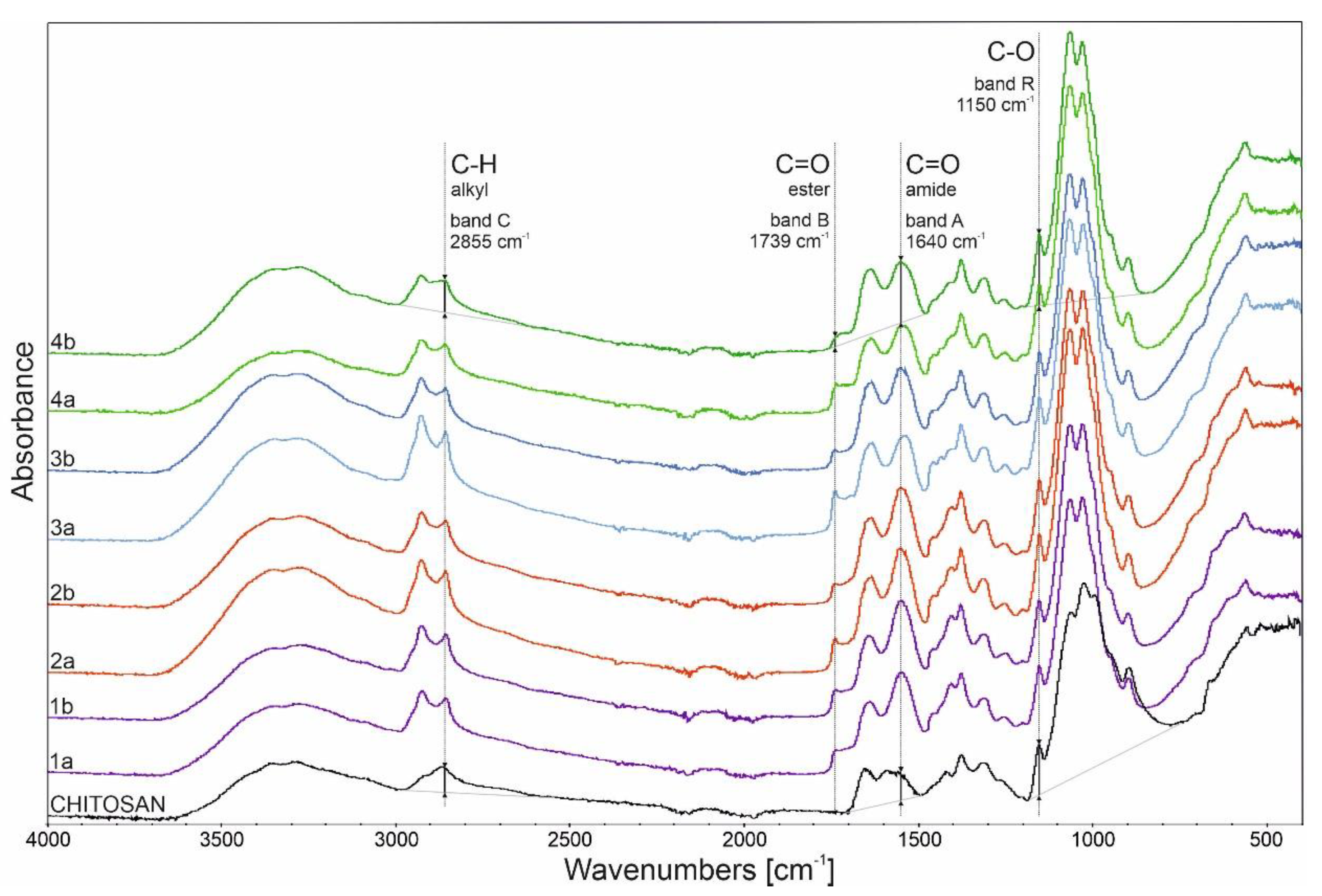
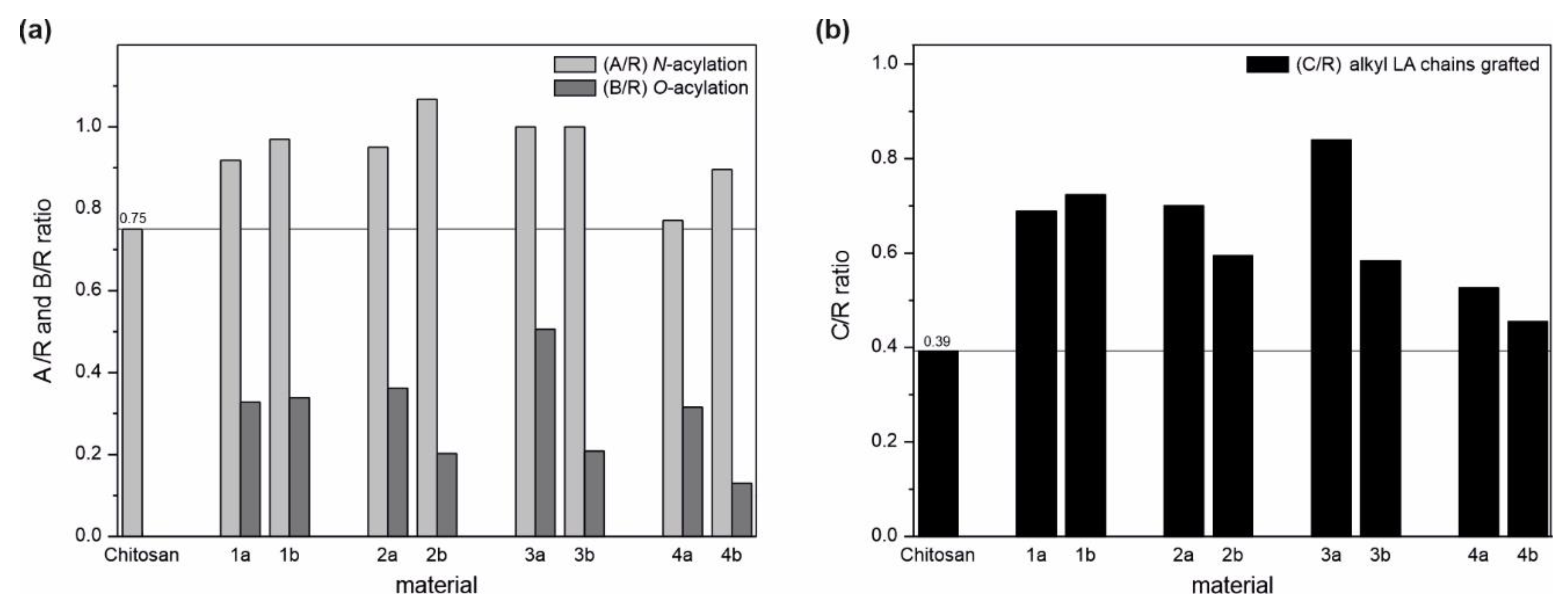
2.1.1. FTIR Analysis
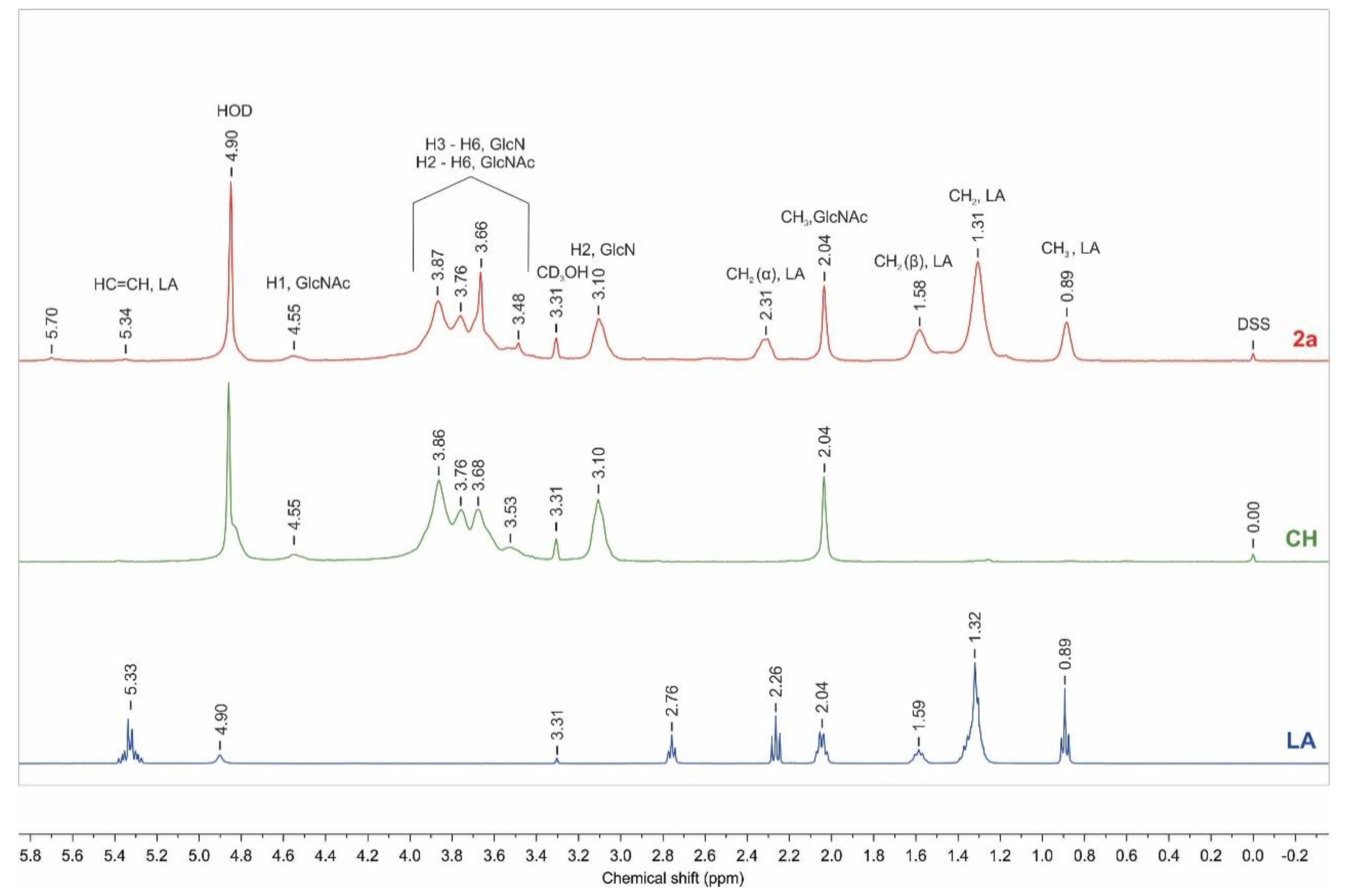
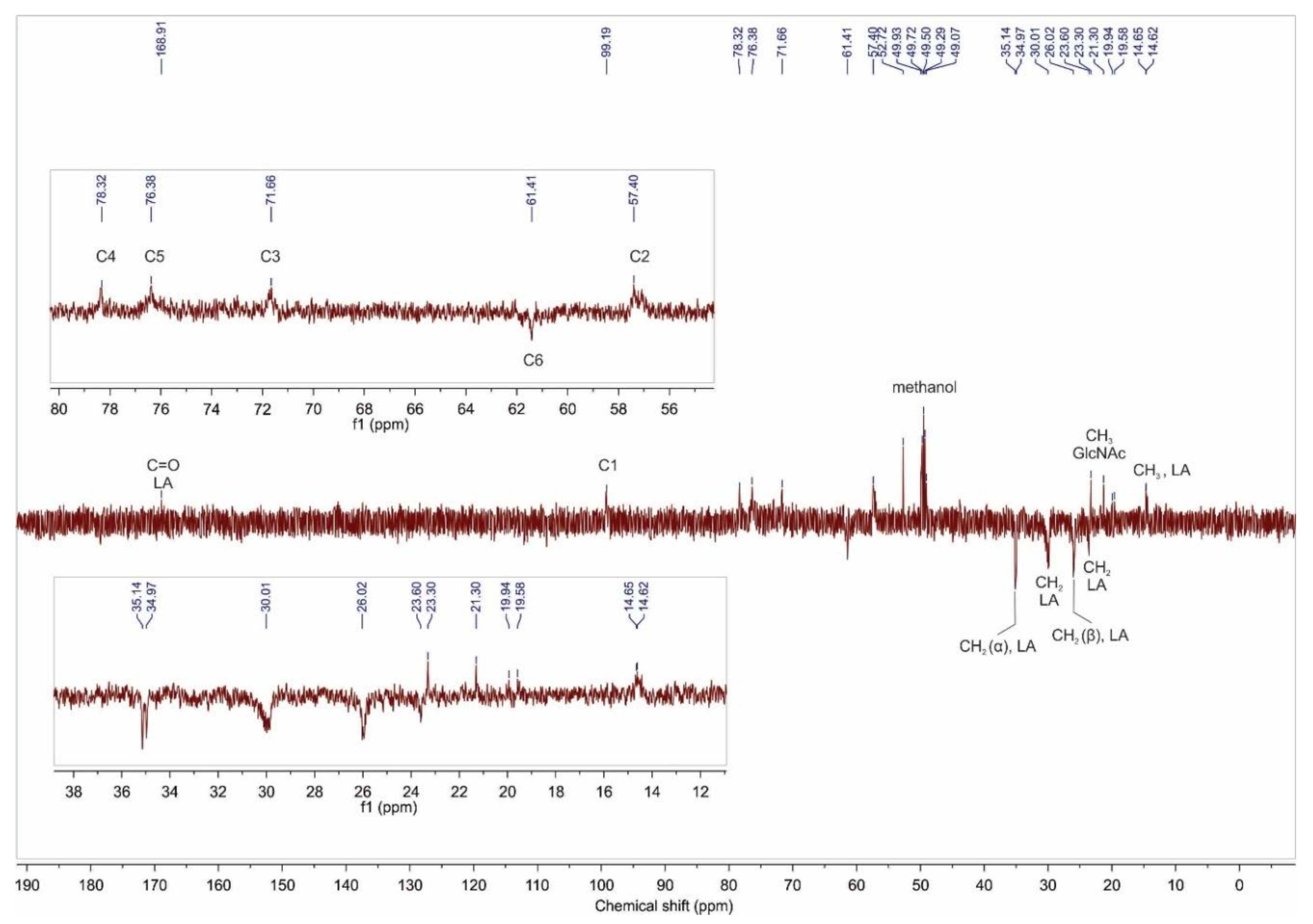
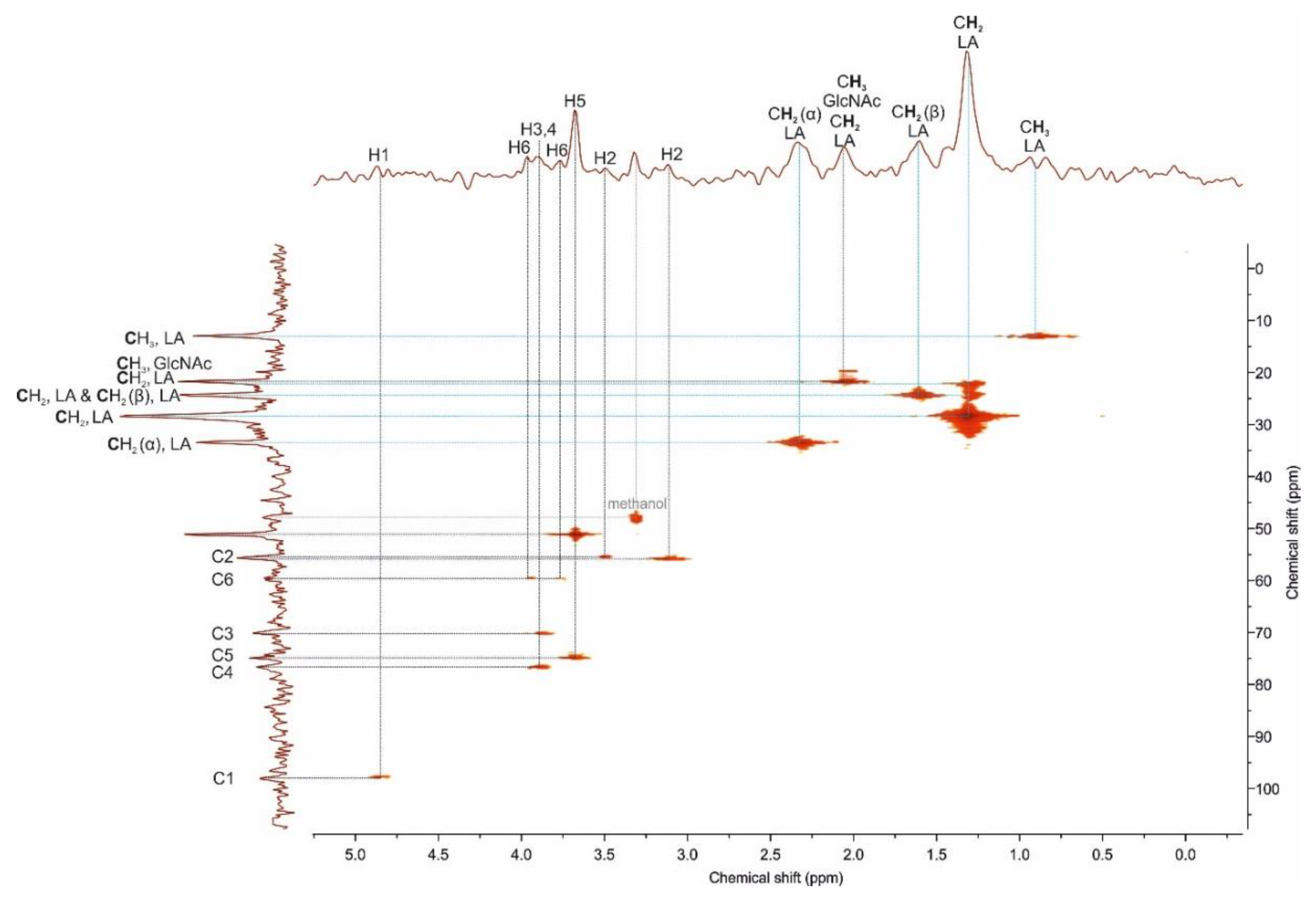
2.1.2. NMR Analysis
2.2. Molecular Weight Determination
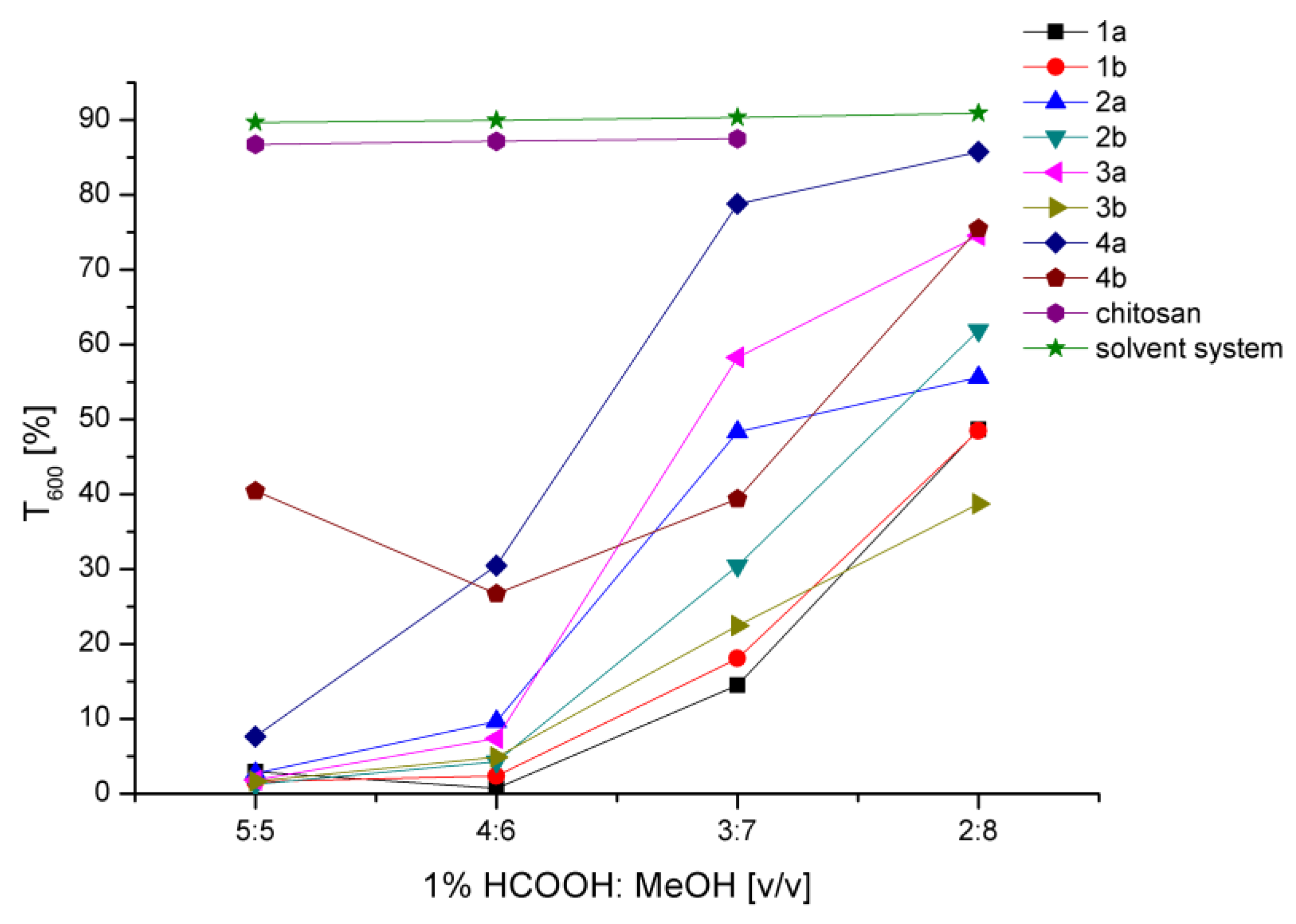
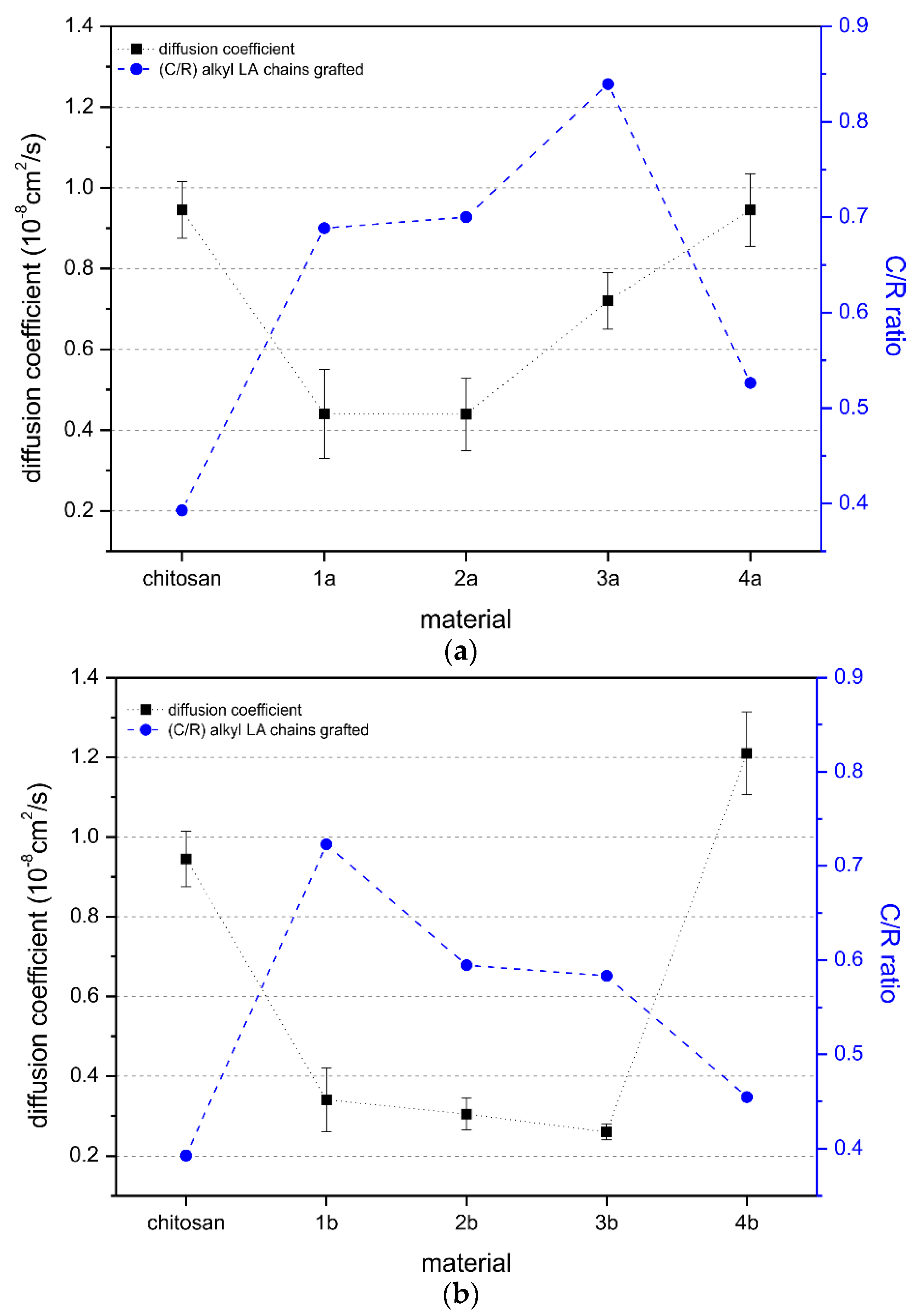
2.3. Behavior in Solution
3. Materials and Methods
3.1. General Information
3.2. Synthesis of Chitosan Derivatives
3.3. Solubility Test
3.4. Fourier Transform Infrared Spectroscopy
3.5. Nuclear Magnetic Resonance Spectroscopy
3.6. Dynamic Light Scattering
3.7. Gel Filtration Chromatography
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Shariatinia, Z. Pharmaceutical applications of chitosan. Adv. Colloid Interface Sci. 2019, 263, 131–194. [Google Scholar] [CrossRef] [PubMed]
- Pellá, M.C.G.; Lima-Tenório, M.K.; Tenório-Neto, E.T.; Guilherme, M.R.; Muniz, E.C.; Rubira, A.F. Chitosan-based hydrogels: From preparation to biomedical applications. Carbohydr. Polym. 2018, 196, 233–245. [Google Scholar] [CrossRef]
- Mohammadzadeh Pakdel, P.; Peighambardoust, S.J. Review on recent progress in chitosan-based hydrogels for wastewater treatment application. Carbohydr. Polym. 2018, 201, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tan, W.; Wei, L.; Chen, Y.; Mi, Y.; Sun, X.; Li, Q.; Dong, F.; Guo, Z. Synthesis of urea-functionalized chitosan derivatives for potential antifungal and antioxidant applications. Carbohydr. Polym. 2019, 215, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Zargar, V.; Asghari, M.; Dashti, A. A Review on Chitin and Chitosan Polymers: Structure, Chemistry, Solubility, Derivatives, and Applications. ChemBioEng Rev. 2015, 2, 204–226. [Google Scholar]
- Pa, J.-H.; Yu, T.L. Light Scattering Study of Chitosan in Acetic Acid Aqueous Solutions. Macromol. Chem. Phys. 2001, 202, 985–991. [Google Scholar] [CrossRef]
- Rinaudo, M.; Pavlov, G.; Desbrie, J.; Desbrières, J. Influence of acetic acid concentration on the solubilization of chitosan. Polymer (Guildf) 1999, 40, 7029–7032. [Google Scholar] [CrossRef]
- Doi, M. Soft Matter Physics, 1st ed.; Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- Bartkowiak, A. Effect of the ionic strength on properties of binary alginate/oligochitosan microcapsules. Colloids Surfaces A Physicochem. Eng. Asp. 2002, 204, 117–124. [Google Scholar] [CrossRef]
- Sæther, H.V.; Holme, H.K.; Maurstad, G.; Smidsrød, O.; Stokke, B.T. Polyelectrolyte complex formation using alginate and chitosan. Carbohydr. Polym. 2008, 74, 813–821. [Google Scholar] [CrossRef]
- Guzmán, J.; Saucedo, I.; Navarro, R.; Revilla, J.; Guibal, E. Vanadium interactions with chitosan: Influence of polymer protonation and metal speciation. Langmuir 2002, 18, 1567–1573. [Google Scholar] [CrossRef]
- Chassary, P.; Vincent, T.; Guibal, E. Metal anion sorption on chitosan and derivative materials: A strategy for polymer modification and optimum use. React. Funct. Polym. 2004, 60, 137–149. [Google Scholar] [CrossRef]
- Sashiwa, H.; Kawasaki, N.; Nakayama, A.; Muraki, E.; Yamamoto, N.; Aiba, S.I. Chemical modification of chitosan. 14:1 synthesis of water-soluble chitosan derivatives by simple acetylation. Biomacromolecules 2002, 3, 1126–1128. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Du, Y.; Yang, J.; Tang, Y.; Li, J.; Wang, X. Self-aggregation and antibacterial activity of N-acylated chitosan. Polymer (Guildf) 2007, 48, 3098–3106. [Google Scholar] [CrossRef]
- Hirano, S.; Yamaguchi, Y.; Kamiya, M. Novel N-saturated-fatty-acyl derivatives of chitosan soluble in water and in aqueous acid and alkaline solutions. Carbohydr. Polym. 2002, 48, 203–207. [Google Scholar] [CrossRef]
- Jothimani, B.; Sureshkumar, S.; Venkatachalapathy, B. Hydrophobic structural modification of chitosan and its impact on nanoparticle synthesis—A physicochemical study. Carbohydr. Polym. 2017, 173, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Kurita, K.; Ikeda, H.; Yoshida, Y.; Shimojoh, M.; Harata, M. Chemoselective Protection of the Amino Groups of Chitosan by Controlled Phthaloylation: Facile Preparation of a Precursor Useful for Chemical Modifications. Biomacromolecules 2002, 3, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kurita, K.; Ikeda, H.; Shimojoh, M.; Yang, J. N-Phthaloylated Chitosan as an Essential Precursor for Controlled Chemical Modifications of Chitosan: Synthesis and Evaluation. Polym. J. 2007, 39, 945–952. [Google Scholar] [CrossRef]
- Badawy, M.E.I.; Rabea, E.I.; Rogge, T.M.; Stevens, C.V.; Steurbaut, W.; Höfte, M.; Smagghe, G. Fungicidal and Insecticidal Activity of O-Acyl Chitosan Derivatives. Polym. Bull. 2005, 54, 279–289. [Google Scholar] [CrossRef]
- Badawy, M.E.I.; Rabea, E.I.; Rogge, T.M.; Stevens, C.V.; Smagghe, G.; Steurbaut, W.; Höfte, M. Synthesis and fungicidal activity of new N,O-acyl chitosan derivatives. Biomacromolecules 2004, 5, 589–595. [Google Scholar] [CrossRef]
- Totaro, K.A.; Liao, X.; Bhattacharya, K.; Finneman, J.I.; Sperry, J.B.; Massa, M.A.; Thorn, J.; Ho, S.V.; Pentelute, B.L. Systematic Investigation of EDC/sNHS-Mediated Bioconjugation Reactions for Carboxylated Peptide Substrates. Bioconjug. Chem. 2016, 27, 994–1004. [Google Scholar] [CrossRef]
- Nam, K.; Kimura, T.; Kishida, A. Controlling coupling reaction of EDC and NHS for preparation of collagen gels using ethanol/water co-solvents. Macromol. Biosci. 2008, 8, 32–37. [Google Scholar] [CrossRef]
- Vashist, S.K. Comparison of 1-Ethyl-3-(3-Dimethylaminopropyl) Carbodiimide Based Strategies to Crosslink Antibodies on Amine-Functionalized Platforms for Immunodiagnostic Applications. Diagnostics 2012, 2, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Singh, J. Synthesis and Characterization of Fatty Acid Grafted Chitosan Polymer and Their Nanomicelles for Nonviral Gene Delivery Applications. Bioconjug. Chem. 2017, 28, 2772–2783. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Jo, W.H.; Kwon, I.C.; Kim, Y.H.; Jeong, S.Y. Structural determination and interior polarity of self-aggregates prepared from deoxycholic acid-modified chitosan in water. Macromolecules 1998, 31, 378–383. [Google Scholar] [CrossRef]
- Casettari, L.; Vllasaliu, D.; Mantovani, G.; Howdle, S.M.; Stolnik, S.; Illum, L. Effect of PEGylation on the Toxicity and Permeability Enhancement of Chitosan. Biomacromolecules 2010, 11, 2854–2865. [Google Scholar] [CrossRef]
- Everaerts, F.; Torrianni, M.; Hendriks, M.; Feijen, J. Biomechanical properties of carbodiimide crosslinked collagen: Influence of the formation of ester crosslinks. J. Biomed. Mater. Res. Part A 2008, 85A, 547–555. [Google Scholar] [CrossRef]
- Dang, Q.; Zhang, Q.; Liu, C.; Yan, J.; Chang, G.; Xin, Y.; Cheng, X.; Cao, Y.; Gao, H.; Liu, Y. Decanoic acid functionalized chitosan: Synthesis, characterization, and evaluation as potential wound dressing material. Int. J. Biol. Macromol. 2019, 139, 1046–1053. [Google Scholar] [CrossRef]
- Nakajima, N.; Ikada, Y. Mechanism of Amide Formation by Carbodiimide for Bioconjugation in Aqueous Media. Bioconjug. Chem. 1995, 6, 123–130. [Google Scholar] [CrossRef]
- Liu, C.-G.; Goud, H.; Desai, K.; Chen, X.-G.; Park, H.-J. Preparation and Characterization of Nanoparticles Containing Trypsin Based on Hydrophobically Modified Chitosan. J. Agric. Food Chem. 2005, 53, 1728–1733. [Google Scholar] [CrossRef]
- Jiang, G.-B.; Quan, D.; Liao, K.; Wang, H. Novel Polymer Micelles Prepared from Chitosan Grafted Hydrophobic Palmitoyl Groups for Drug Delivery. Mol. Pharm. 2006, 3, 152–160. [Google Scholar] [CrossRef]
- Sam, S.; Touahir, L.; Salvador Andresa, J.; Allongue, P.; Chazalviel, J.-N.; Gouget-Laemmel, A.C.; Henry de Villeneuve, C.; Moraillon, A.; Ozanam, F.; Gabouze, N.; et al. Semiquantitative Study of the EDC/NHS Activation of Acid Terminal Groups at Modified Porous Silicon Surfaces. Langmuir 2010, 26, 809–814. [Google Scholar] [CrossRef]
- Wang, C.; Yan, Q.; Liu, H.-B.; Zhou, X.-H.; Xiao, S.-J. Different EDC/NHS Activation Mechanisms between PAA and PMAA Brushes and the Following Amidation Reactions. Langmuir 2011, 27, 12058–12068. [Google Scholar] [CrossRef] [PubMed]
- Niemczyk, A.; Kmieciak, A.; El Fray, M.; Piegat, A. The influence of C18-fatty acids on chemical structure of chitosan derivatives and their thermal properties. Prog. Chem. Appl. Chitin its Deriv. 2016, 21, 165–175. [Google Scholar] [CrossRef][Green Version]
- Ortona, O.; Errico, G.D.; Mangiapia, G.; Ciccarelli, D. The aggregative behavior of hydrophobically modified chitosans with high substitution degree in aqueous solution. Carbohydr. Polym. 2008, 74, 16–22. [Google Scholar] [CrossRef]
- Esquenet, C.; Terech, P.; Boué, F.; Buhler, E. Structural and rheological properties of hydrophobically modified polysaccharide associative networks. Langmuir 2004, 20, 3583–3592. [Google Scholar] [CrossRef]
- Sogias, I.A.; Khutoryanskiy, V.V.; Williams, A.C. Exploring the factors affecting the solubility of chitosan in water. Macromol. Chem. Phys. 2010, 211, 426–433. [Google Scholar] [CrossRef]
- Kubota, N.; Tatsumoto, N.; Sano, T.; Toya, K. A simple preparation of half N-acetylated chitosan highly soluble in water and aqueous organic solvents. Carbohydr. Res. 2000, 324, 268–274. [Google Scholar] [CrossRef]
- Qin, C.; Li, H.; Xiao, Q.; Liu, Y.; Zhu, J.; Du, Y. Water-solubility of chitosan and its antimicrobial activity. Carbohydr. Polym. 2006, 63, 367–374. [Google Scholar] [CrossRef]
- Jeong, Y.I.; Kim, D.G.; Jang, M.K.; Nah, J.W. Preparation and spectroscopic characterization of methoxy poly(ethylene glycol)-grafted water-soluble chitosan. Carbohydr. Res. 2008, 343, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Buhler, E.; Rinaudo, M. Structural and Dynamical Properties of Semirigid Polyelectrolyte Solutions: A Light-Scattering Study. Macromolecules 2000, 33, 2098–2106. [Google Scholar] [CrossRef]
- Terbojevich, M.; Cosani, A.; Conio, G.; Marsano, E.; Bianchi, E. Chitosan: chain rigidity and mesophase formation. Carbohydr. Res. 1991, 209, 251–260. [Google Scholar] [CrossRef]
- Errington, N.; Harding, S.E.; Vårum, K.M.; Illum, L. Hydrodynamic characterization of chitosans varying in degree of acetylation. Int. J. Biol. Macromol. 1993, 15, 113–117. [Google Scholar] [CrossRef]
- Stetefeld, J.; McKenna, S.A.; Patel, T.R. Dynamic light scattering: a practical guide and applications in biomedical sciences. Biophys. Rev. 2016, 8, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Philippova, O.E.; Volkov, E.V.; Sitnikova, N.L.; Khokhlov, A.R.; Desbrieres, J.; Rinaudo, M. Two Types of Hydrophobic Aggregates in Aqueous Solutions of Chitosan and Its Hydrophobic Derivative. Biomacromolecules 2001, 2, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Chen, A.; Johnson, C.S. An Improved Diffusion-Ordered Spectroscopy Experiment Incorporating Bipolar-Gradient Pulses. J. Magn. Reson. 1995, 115, 260–264. [Google Scholar] [CrossRef]
Sample Availability: Not available. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Number-Average Molecular Weight (Mn) [kDa] | Weight-Average Molecular Weight (Mw) [kDa] | Polydispersity Index (Mw/Mn) |
|---|---|---|---|
| chitosan | 19.9 | 145.7 | 7.3 |
| 1a | 13.2 | 53.0 | 4.0 |
| 1b | 15.1 | 68.4 | 4.5 |
| 2a | 15.1 | 62.0 | 4.1 |
| 2b | 12.0 | 69.9 | 5.8 |
| 3a | 13.6 | 65.8 | 4.8 |
| 3b | 12.8 | 63.8 | 4.9 |
| 4a | 13.2 | 66.5 | 5.0 |
| 4b | 19.5 | 145.8 | 7.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piegat, A.; Goszczyńska, A.; Idzik, T.; Niemczyk, A. The Importance of Reaction Conditions on the Chemical Structure of N,O-Acylated Chitosan Derivatives. Molecules 2019, 24, 3047. https://doi.org/10.3390/molecules24173047
Piegat A, Goszczyńska A, Idzik T, Niemczyk A. The Importance of Reaction Conditions on the Chemical Structure of N,O-Acylated Chitosan Derivatives. Molecules. 2019; 24(17):3047. https://doi.org/10.3390/molecules24173047
Chicago/Turabian StylePiegat, Agnieszka, Agata Goszczyńska, Tomasz Idzik, and Agata Niemczyk. 2019. "The Importance of Reaction Conditions on the Chemical Structure of N,O-Acylated Chitosan Derivatives" Molecules 24, no. 17: 3047. https://doi.org/10.3390/molecules24173047
APA StylePiegat, A., Goszczyńska, A., Idzik, T., & Niemczyk, A. (2019). The Importance of Reaction Conditions on the Chemical Structure of N,O-Acylated Chitosan Derivatives. Molecules, 24(17), 3047. https://doi.org/10.3390/molecules24173047