
RNA Secondary Structure-Based Design of Antisense Peptide Nucleic Acids for Modulating Disease-Associated Aberrant Tau Pre-mRNA Alternative Splicing

 and
and
Abstract
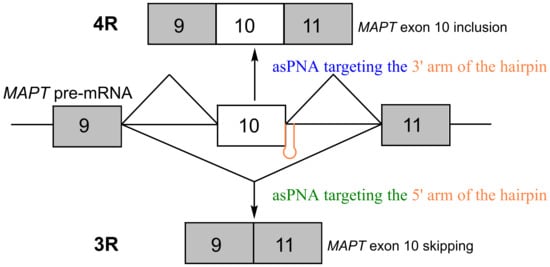
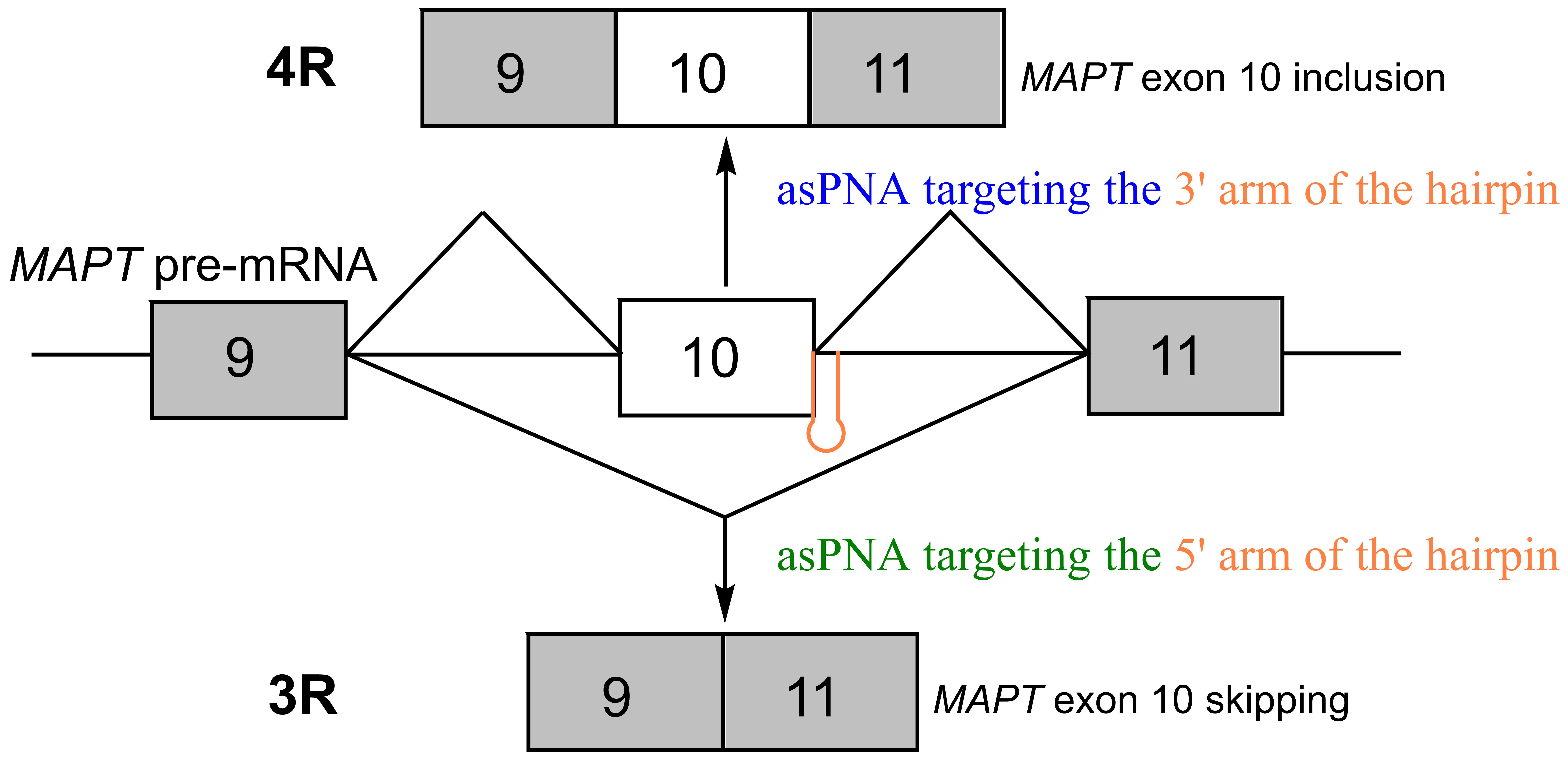
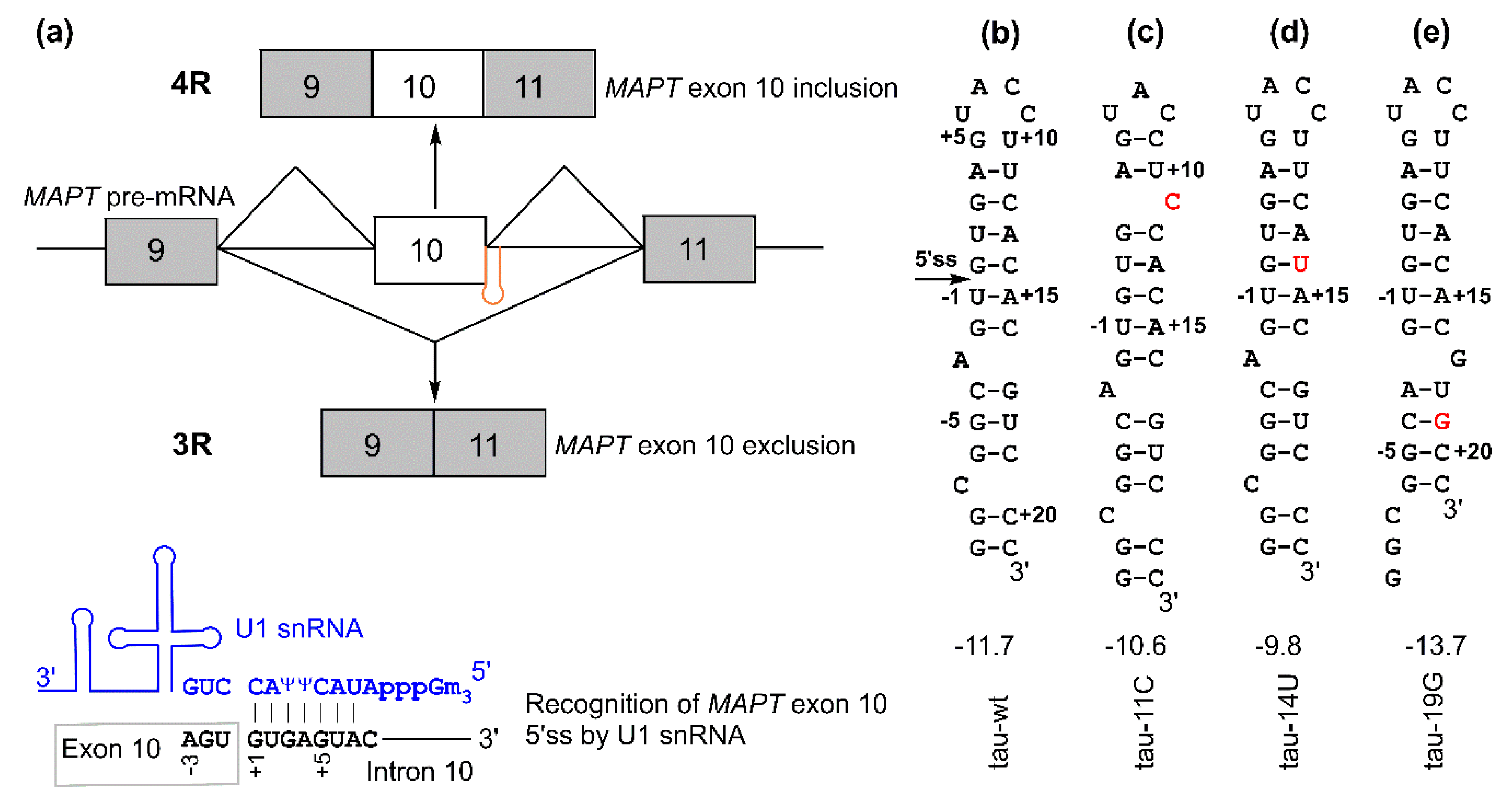
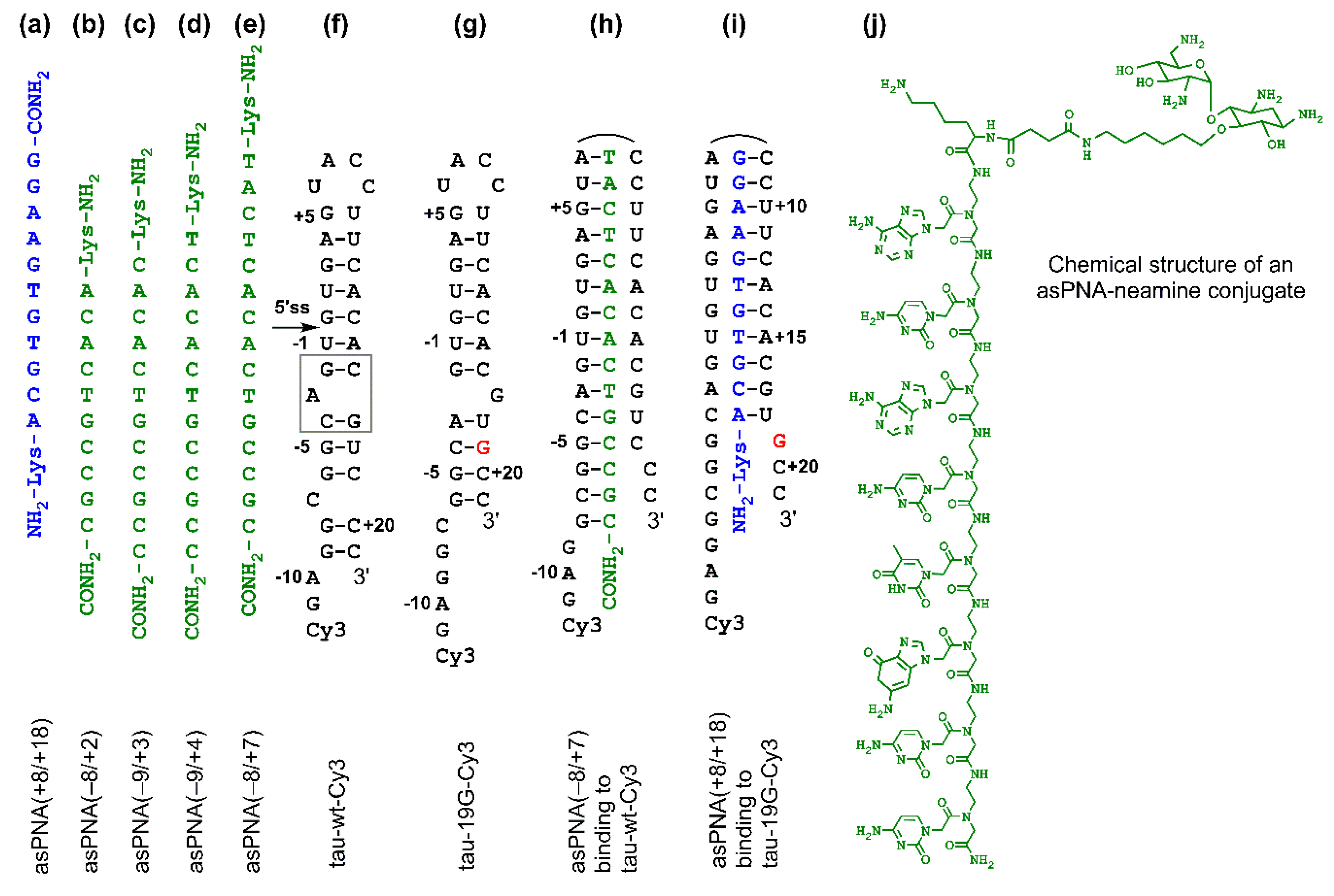
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
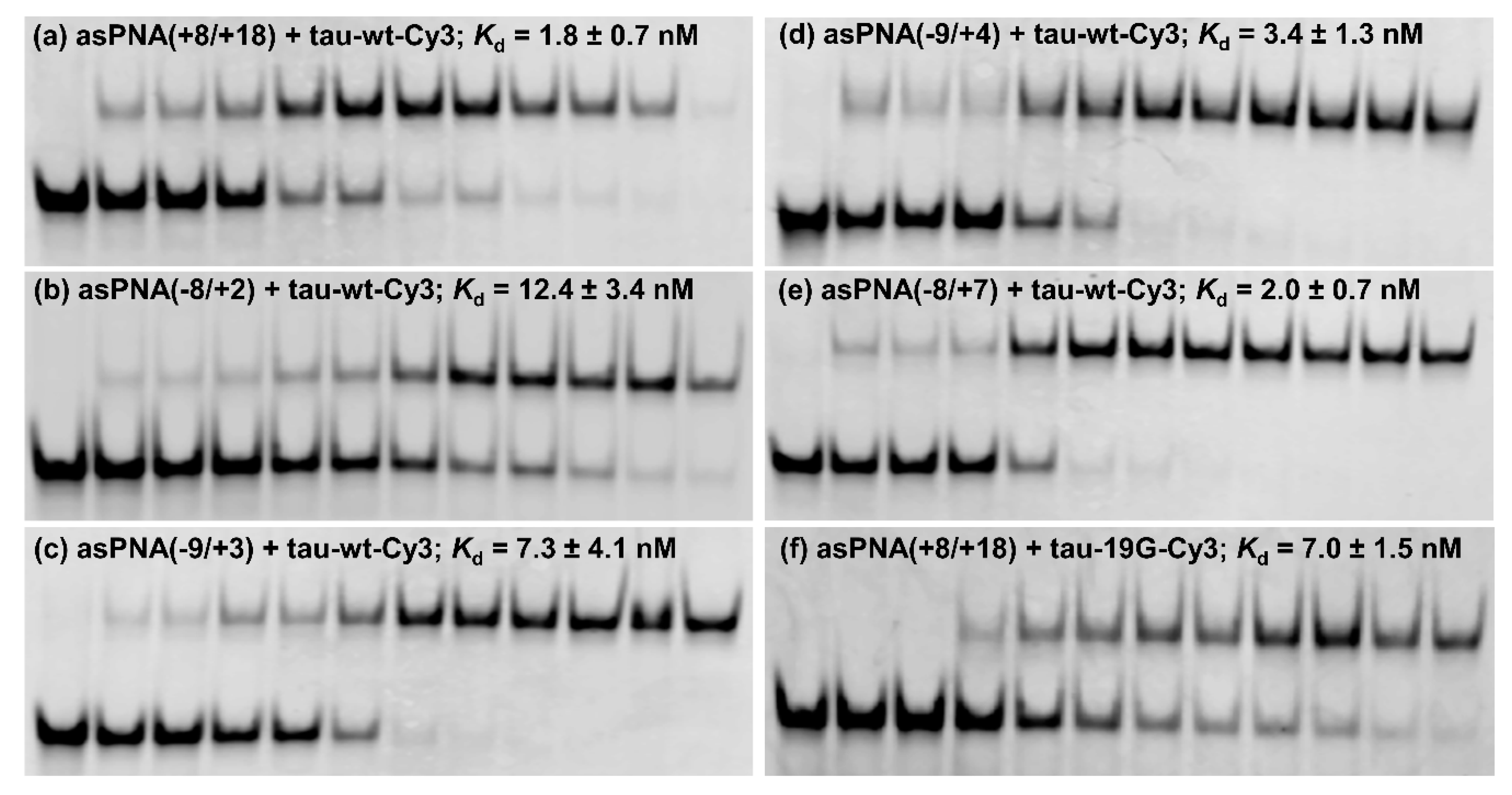
2.1. asPNAs Can Invade Tau Pre-mRNA Hairpin
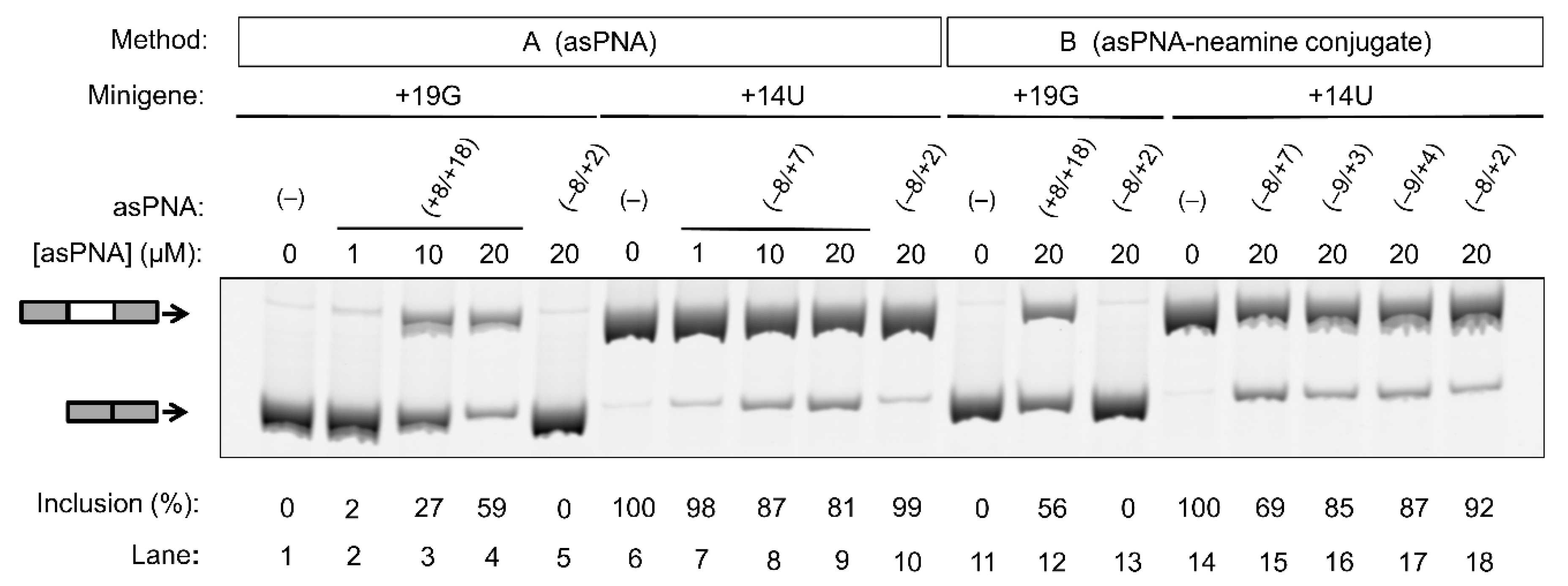
2.2. asPNAs Can Alter Tau Minigene Pre-mRNA Splicing in Cell Cultures
3. Materials and Methods
3.1. General Methods and Synthesis of PNA Oligomers
3.2. Nondenaturing Polyacrylamide Gel Electrophoresis
3.3. Cell Culture Minigene Splicing Assay
3.4. Real-Time PCR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Irwin, D.J. Tauopathies as clinicopathological entities. Parkinsonism Relat. Disord. 2016, 22 (Suppl. 1), S29–S33. [Google Scholar] [CrossRef]
- Kosik, K.S.; Shimura, H. Phosphorylated tau and the neurodegenerative foldopathies. Biochim. Biophys. Acta 2005, 1739, 298–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.; Zhang, W.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from Pick′s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer′s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Wszolek, Z.K.; Tsuboi, Y.; Ghetti, B.; Pickering-Brown, S.; Baba, Y.; Cheshire, W.P. Frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Orphanet J. Rare Dis. 2006, 1, 30. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Gotz, J. Tau-based therapies in neurodegeneration: Opportunities and challenges. Nat. Rev. Drug Discov. 2017, 16, 863–883. [Google Scholar] [CrossRef]
- D′Souza, I.; Poorkaj, P.; Hong, M.; Nochlin, D.; Lee, V.M.; Bird, T.D.; Schellenberg, G.D. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc. Natl. Acad. Sci. USA 1999, 96, 5598–5603. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Zhukareva, V.; Vogelsberg-Ragaglia, V.; Wszolek, Z.; Reed, L.; Miller, B.I.; Geschwind, D.H.; Bird, T.D.; McKeel, D.; Goate, A.; et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 1998, 282, 1914–1917. [Google Scholar] [CrossRef]
- Hirokawa, N. Microtubule organization and dynamics dependent on microtubule-associated proteins. Curr. Opin. Cell Biol. 1994, 6, 74–81. [Google Scholar] [CrossRef]
- Kellogg, E.H.; Hejab, N.M.A.; Poepsel, S.; Downing, K.H.; DiMaio, F.; Nogales, E. Near-atomic model of microtubule-tau interactions. Science 2018, 360, 1242–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wszolek, Z.K.; Slowinski, J.; Golan, M.; Dickson, D.W. Frontotemporal dementia and parkinsonism linked to chromosome 17. Folia Neuropathol. 2005, 43, 258–270. [Google Scholar] [PubMed]
- Young, J.J.; Lavakumar, M.; Tampi, D.; Balachandran, S.; Tampi, R.R. Frontotemporal dementia: Latest evidence and clinical implications. Ther. Adv. Psychopharmacol. 2018, 8, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Ghetti, B.; Oblak, A.L.; Boeve, B.F.; Johnson, K.A.; Dickerson, B.C.; Goedert, M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: A chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 2015, 41, 24–46. [Google Scholar] [CrossRef] [PubMed]
- Stanford, P.M.; Shepherd, C.E.; Halliday, G.M.; Brooks, W.S.; Schofield, P.W.; Brodaty, H.; Martins, R.N.; Kwok, J.B.J.; Schofield, P.R. Mutations in the tau gene that cause an increase in three repeat tau and frontotemporal dementia. Brain 2003, 126, 814–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoch, K.M.; DeVos, S.L.; Miller, R.L.; Chun, S.J.; Norrbom, M.; Wozniak, D.F.; Dawson, H.N.; Bennett, C.F.; Rigo, F.; Miller, T.M. Increased 4R-Tau Induces Pathological Changes in a Human-Tau Mouse Model. Neuron 2016, 90, 941–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dregni, A.J.; Mandala, V.S.; Wu, H.; Elkins, M.R.; Wang, H.K.; Hung, I.; DeGrado, W.F.; Hong, M. In vitro 0N4R tau fibrils contain a monomorphic beta-sheet core enclosed by dynamically heterogeneous fuzzy coat segments. Proc. Natl. Acad. Sci. USA 2019, 116, 16357–16366. [Google Scholar] [CrossRef]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef]
- McCarthy, A.; Lonergan, R.; Olszewska, D.A.; O′Dowd, S.; Cummins, G.; Magennis, B.; Fallon, E.M.; Pender, N.; Huey, E.D.; Cosentino, S.; et al. Closing the tau loop: The missing tau mutation. Brain 2015, 138, 3100–3109. [Google Scholar] [CrossRef]
- Donahue, C.P.; Muratore, C.; Wu, J.Y.; Kosik, K.S.; Wolfe, M.S. Stabilization of the tau exon 10 stem loop alters pre-mRNA splicing. J. Biol. Chem. 2006, 281, 23302–23306. [Google Scholar] [CrossRef]
- Varani, L.; Hasegawa, M.; Spillantini, M.G.; Smith, M.J.; Murrell, J.R.; Ghetti, B.; Klug, A.; Goedert, M.; Varani, G. Structure of tau exon 10 splicing regulatory element RNA and destabilization by mutations of frontotemporal dementia and parkinsonism linked to chromosome 17. Proc. Natl. Acad. Sci. USA 1999, 96, 8229–8234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buratti, E.; Baralle, F.E. Influence of RNA secondary structure on the pre-mRNA splicing process. Mol. Cell Biol. 2004, 24, 10505–10514. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.; Houlden, H.; Baker, M.; Adamson, J.; Lewis, J.; Prihar, G.; Pickering-Brown, S.; Duff, K.; Hutton, M. 5′ splice site mutations in tau associated with the inherited dementia FTDP-17 affect a stem-loop structure that regulates alternative splicing of exon 10. J. Biol. Chem. 1999, 274, 15134–15143. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.; Moss, W.N.; Spencer, A.; Zhang, P.; Childs-Disney, J.L.; Disney, M.D. The RNA encoding the microtubule-associated protein tau has extensive structure that affects its biology. PLoS ONE 2019, 14, e0219210. [Google Scholar] [CrossRef] [PubMed]
- Roca, X.; Krainer, A.R.; Eperon, I.C. Pick one, but be quick: 5′ splice sites and the problems of too many choices. Genes Dev. 2013, 27, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Roca, X.; Akerman, M.; Gaus, H.; Berdeja, A.; Bennett, C.F.; Krainer, A.R. Widespread recognition of 5′ splice sites by noncanonical base-pairing to U1 snRNA involving bulged nucleotides. Genes Dev. 2012, 26, 1098–1109. [Google Scholar] [CrossRef]
- Tan, J.; Ho, J.; Zhong, Z.; Luo, S.; Chen, G.; Roca, X. Noncanonical registers and base pairs in human 5′ splice-site selection. Nucleic Acids Res. 2016, 44, 3908–3921. [Google Scholar] [CrossRef]
- Zhou, J.; Yu, Q.; Zou, T. Alternative splicing of exon 10 in the tau gene as a target for treatment of tauopathies. BMC Neurosci. 2008, 9 (Suppl. 2), S10. [Google Scholar]
- Tan, J.; Yang, L.; Ong, A.A.L.; Shi, J.; Zhong, Z.; Lye, M.L.; Liu, S.; Lisowiec-Wachnicka, J.; Kierzek, R.; Roca, X.; et al. A disease-causing intronic point mutation C19G alters tau exon 10 splicing via RNA secondary structure rearrangement. Biochemistry 2019, 58, 1565–1578. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.H.; Disney, M.D.; Childs, J.L.; Schroeder, S.J.; Zuker, M.; Turner, D.H. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. USA 2004, 101, 7287–7292. [Google Scholar] [CrossRef] [Green Version]
- Qian, W.; Liu, F. Regulation of alternative splicing of tau exon 10. Neurosci. Bull. 2014, 30, 367–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D′Souza, I.; Schellenberg, G.D. Tau Exon 10 expression involves a bipartite intron 10 regulatory sequence and weak 5′ and 3′ splice sites. J. Biol. Chem. 2002, 277, 26587–26599. [Google Scholar] [CrossRef] [PubMed]
- Lisowiec, J.; Magner, D.; Kierzek, E.; Lenartowicz, E.; Kierzek, R. Structural determinants for alternative splicing regulation of the MAPT pre-mRNA. RNA Biol. 2015, 12, 330–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Gao, Q.S.; Wang, Y.; Lafyatis, R.; Stamm, S.; Andreadis, A. Tau exon 10, whose missplicing causes frontotemporal dementia, is regulated by an intricate interplay of cis elements and trans factors. J. Neurochem. 2004, 88, 1078–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600. [Google Scholar] [CrossRef]
- Kalbfuss, B.; Mabon, S.A.; Misteli, T. Correction of alternative splicing of tau in frontotemporal dementia and parkinsonism linked to chromosome 17. J. Biol. Chem. 2001, 276, 42986–42993. [Google Scholar] [CrossRef]
- Sazani, P.; Kole, R. Therapeutic potential of antisense oligonucleotides as modulators of alternative splicing. J. Clin. Invest. 2003, 112, 481–486. [Google Scholar] [CrossRef] [Green Version]
- Rigo, F.; Hua, Y.; Chun, S.J.; Prakash, T.P.; Krainer, A.R.; Bennett, C.F. Synthetic oligonucleotides recruit ILF2/3 to RNA transcripts to modulate splicing. Nat. Chem. Biol. 2012, 8, 555–561. [Google Scholar] [CrossRef] [Green Version]
- Havens, M.A.; Duelli, D.M.; Hastings, M.L. Targeting RNA splicing for disease therapy. Wiley Interdiscip. Rev. RNA 2013, 4, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Sud, R.; Geller, E.T.; Schellenberg, G.D. Antisense-mediated exon skipping decreases tau protein expression: A potential therapy for tauopathies. Mol. Ther. Nucleic Acids 2014, 3, e180. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Hyrup, B.; Nielsen, P.E. Peptide nucleic acids (PNA): Synthesis, properties and potential applications. Bioorg. Med. Chem. 1996, 4, 5–23. [Google Scholar] [CrossRef]
- Egholm, M.; Nielsen, P.E.; Buchardt, O.; Berg, R.H. Recognition of guanine and adenine in DNA by cytosine and thymine containing peptide nucleic acids (PNA). J. Am. Chem. Soc. 1992, 114, 9677–9678. [Google Scholar] [CrossRef]
- Egholm, M.; Buchardt, O.; Nielsen, P.E.; Berg, R.H. Peptide nucleic acids (PNA). Oligonucleotide analogs with an achiral peptide backbone. J. Am. Chem. Soc. 1992, 114, 1895–1897. [Google Scholar] [CrossRef]
- Egholm, M.; Buchardt, O.; Christensen, L.; Behrens, C.; Freier, S.M.; Driver, D.A.; Berg, R.H.; Kim, S.K.; Norden, B.; Nielsen, P.E. PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules. Nature 1993, 365, 566–568. [Google Scholar] [CrossRef]
- Armitage, B.A. The impact of nucleic acid secondary structure on PNA hybridization. Drug Discov. Today 2003, 8, 222–228. [Google Scholar] [CrossRef]
- Demidov, V.; Frank-Kamenetskii, M.D.; Egholm, M.; Buchardt, O.; Nielsen, P.E. Sequence selective double strand DNA cleavage by peptide nucleic acid (PNA) targeting using nuclease S1. Nucleic Acids Res. 1993, 21, 2103–2107. [Google Scholar] [CrossRef]
- Winssinger, N.; Damoiseaux, R.; Tully, D.C.; Geierstanger, B.H.; Burdick, K.; Harris, J.L. PNA-encoded protease substrate microarrays. Chem. Biol. 2004, 11, 1351–1360. [Google Scholar] [CrossRef]
- Komiyama, M.; Ye, S.; Liang, X.; Yamamoto, Y.; Tomita, T.; Zhou, J.M.; Aburatani, H. PNA for one-base differentiating protection of DNA from nuclease and its use for SNPs detection. J. Am. Chem. Soc. 2003, 125, 3758–3762. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, A.; Ponzio, N.M.; Pandey, V.N. Immunological response to peptide nucleic acid and its peptide conjugate targeted to transactivation response (TAR) region of HIV-1 RNA genome. Oligonucleotides 2008, 18, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Hanvey, J.C.; Peffer, N.J.; Bisi, J.E.; Thomson, S.A.; Cadilla, R.; Josey, J.A.; Ricca, D.J.; Hassman, C.F.; Bonham, M.A.; Au, K.G.; et al. Antisense and antigene properties of peptide nucleic acids. Science 1992, 258, 1481–1485. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.Y.; Ju, Y.; Park, H. A highly effective and long-lasting inhibition of miRNAs with PNA-based antisense oligonucleotides. Mol. Cells 2009, 28, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Abes, S.; Turner, J.J.; Ivanova, G.D.; Owen, D.; Williams, D.; Arzumanov, A.; Clair, P.; Gait, M.J.; Lebleu, B. Efficient splicing correction by PNA conjugation to an R6-Penetratin delivery peptide. Nucleic Acids Res. 2007, 35, 4495–4502. [Google Scholar] [CrossRef] [PubMed]
- Das, I.; Desire, J.; Manvar, D.; Baussanne, I.; Pandey, V.N.; Decout, J.L. A peptide nucleic acid-aminosugar conjugate targeting transactivation response element of HIV-1 RNA genome shows a high bioavailability in human cells and strongly inhibits tat-mediated transactivation of HIV-1 transcription. J. Med. Chem. 2012, 55, 6021–6032. [Google Scholar] [CrossRef]
- Huang, X.W.; Pan, J.; An, X.Y.; Zhuge, H.X. Inhibition of bacterial translation and growth by peptide nucleic acids targeted to domain II of 23S rRNA. J. Pept. Sci. 2007, 13, 220–226. [Google Scholar]
- Fabani, M.M.; Abreu-Goodger, C.; Williams, D.; Lyons, P.A.; Torres, A.G.; Smith, K.G.; Enright, A.J.; Gait, M.J.; Vigorito, E. Efficient inhibition of miR-155 function in vivo by peptide nucleic acids. Nucleic Acids Res. 2010, 38, 4466–4475. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.J.; Bahal, R.; Babar, I.A.; Pincus, Z.; Barrera, F.; Liu, C.; Svoronos, A.; Braddock, D.T.; Glazer, P.M.; Engelman, D.M.; et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature 2015, 518, 107–110. [Google Scholar] [CrossRef]
- Quijano, E.; Bahal, R.; Ricciardi, A.; Saltzman, W.M.; Glazer, P.M. Therapeutic Peptide Nucleic Acids: Principles, Limitations, and Opportunities. Yale J. Biol. Med. 2017, 90, 583–598. [Google Scholar]
- Gupta, A.; Quijano, E.; Liu, Y.; Bahal, R.; Scanlon, S.E.; Song, E.; Hsieh, W.C.; Braddock, D.E.; Ly, D.H.; Saltzman, W.M.; et al. Anti-tumor Activity of miniPEG-γ-Modified PNAs to Inhibit MicroRNA-210 for Cancer Therapy. Mol. Ther. Nucleic Acids 2017, 9, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, A.S.; Bahal, R.; Farrelly, J.S.; Quijano, E.; Bianchi, A.H.; Luks, V.L.; Putman, R.; Lopez-Giraldez, F.; Coskun, S.; Song, E.; et al. In utero nanoparticle delivery for site-specific genome editing. Nat. Commun. 2018, 9, 2481. [Google Scholar] [CrossRef] [PubMed]
- Ozes, A.R.; Wang, Y.; Zong, X.; Fang, F.; Pilrose, J.; Nephew, K.P. Therapeutic targeting using tumor specific peptides inhibits long non-coding RNA HOTAIR activity in ovarian and breast cancer. Sci. Rep. 2017, 7, 894. [Google Scholar] [CrossRef]
- Kolevzon, N.; Nasereddin, A.; Naik, S.; Yavin, E.; Dzikowski, R. Use of peptide nucleic acids to manipulate gene expression in the malaria parasite Plasmodium falciparum. PLoS ONE 2014, 9, e86802. [Google Scholar] [CrossRef]
- Gait, M.J.; Arzumanov, A.A.; McClorey, G.; Godfrey, C.; Betts, C.; Hammond, S.; Wood, M.J.A. Cell-Penetrating Peptide Conjugates of Steric Blocking Oligonucleotides as Therapeutics for Neuromuscular Diseases from a Historical Perspective to Current Prospects of Treatment. Nucleic Acid Ther. 2019, 29, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lohse, J.; Dahl, O.; Nielsen, P.E. Double duplex invasion by peptide nucleic acid: A general principle for sequence-specific targeting of double-stranded DNA. Proc. Natl. Acad. Sci. USA 1999, 96, 11804–11808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolina, I.V.; Demidov, V.V.; Soldatenkov, V.A.; Chasovskikh, S.G.; Frank-Kamenetskii, M.D. End invasion of peptide nucleic acids (PNAs) with mixed-base composition into linear DNA duplexes. Nucleic Acids Res. 2005, 33, e146. [Google Scholar] [CrossRef]
- Hu, J.; Corey, D.R. Inhibiting gene expression with peptide nucleic acid (PNA)--peptide conjugates that target chromosomal DNA. Biochemistry 2007, 46, 7581–7589. [Google Scholar] [CrossRef]
- Wittung, P.; Nielsen, P.; Norden, B. Extended DNA-recognition repertoire of peptide nucleic acid (PNA): PNA-dsDNA triplex formed with cytosine-rich homopyrimidine PNA. Biochemistry 1997, 36, 7973–7979. [Google Scholar] [CrossRef] [PubMed]
- Avitabile, C.; Accardo, A.; Ringhieri, P.; Morelli, G.; Saviano, M.; Montagner, G.; Fabbri, E.; Gallerani, E.; Gambari, R.; Romanelli, A. Incorporation of naed peptide nucleic acids into liposomes leads to fast and efficient delivery. Bioconjugate Chem. 2015, 26, 1533–1541. [Google Scholar] [CrossRef]
- Riguet, E.; Tripathi, S.; Chaubey, B.; Desire, J.; Pandey, V.N.; Decout, J.L. A peptide nucleic acid-neamine conjugate that targets and cleaves HIV-1 TAR RNA inhibits viral replication. J. Med. Chem. 2004, 47, 4806–4809. [Google Scholar] [CrossRef]
- Kesy, J.; Patil, K.M.; Kumar, S.M.; Shu, Z.; Yee, Y.H.; Zimmermann, L.; Ong, A.A.L.; Toh, D.F.K.; Krishna, M.S.; Yang, L.; et al. A short chemically modified dsRNA-binding PNA (dbPNA) inhibits influenza viral replication by targeting viral RNA panhandle structure. Bioconjugate Chem. 2019, 30, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Peacey, E.; Rodriguez, L.; Liu, Y.; Wolfe, M.S. Targeting a pre-mRNA structure with bipartite antisense molecules modulates tau alternative splicing. Nucleic Acids Res. 2012, 40, 9836–9849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devi, G.; Yuan, Z.; Lu, Y.; Zhao, Y.; Chen, G. Incorporation of thio-pseudoisocytosine into triplex-forming peptide nucleic acids for enhanced recognition of RNA duplexes. Nucleic Acids Res. 2014, 42, 4008–4018. [Google Scholar] [CrossRef]
- Toh, D.F.K.; Devi, G.; Patil, K.M.; Qu, Q.; Maraswami, M.; Xiao, Y.; Loh, T.P.; Zhao, Y.; Chen, G. Incorporating a guanidine-modified cytosine base into triplex-forming PNAs for the recognition of a C-G pyrimidine-purine inversion site of an RNA duplex. Nucleic Acids Res. 2016, 44, 9071–9082. [Google Scholar] [CrossRef]
- Ong, A.A.L.; Toh, D.F.K.; Patil, K.M.; Meng, Z.; Yuan, Z.; Krishna, M.S.; Devi, G.; Haruehanroengra, P.; Lu, Y.; Xia, K.; et al. General recognition of U-G, U-A, and C-G pairs by double-stranded RNA-binding PNAs incorporated with an artificial nucleobase. Biochemistry 2019, 58, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Krishna, M.S.; Toh, D.F.K.; Meng, Z.; Ong, A.A.L.; Wang, Z.; Lu, Y.; Xia, K.; Prabakaran, M.; Chen, G. Sequence- and structure-specific probing of RNAs by short nucleobase-modified dsRNA-binding PNAs (dbPNAs) incorporating a fluorescent light-up uracil analog. Anal. Chem. 2019, 91, 5331–5338. [Google Scholar] [CrossRef]
- Patil, K.M.; Toh, D.F.K.; Yuan, Z.; Meng, Z.; Shu, Z.; Zhang, H.; Ong, A.A.L.; Krishna, M.S.; Lu, L.; Lu, Y.; et al. Incorporating uracil and 5-halouracils into short peptide nucleic acids for enhanced recognition of A-U pairs in dsRNAs. Nucleic Acids Res. 2018, 46, 7506–7521. [Google Scholar] [CrossRef]
- Li, M.; Zengeya, T.; Rozners, E. Short peptide nucleic acids bind strongly to homopurine tract of double helical RNA at pH 5.5. J. Am. Chem. Soc. 2010, 132, 8676–8681. [Google Scholar] [CrossRef]
- Gupta, P.; Zengeya, T.; Rozners, E. Triple helical recognition of pyrimidine inversions in polypurine tracts of RNA by nucleobase-modified PNA. Chem. Commun. 2011, 47, 11125–11127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zengeya, T.; Gupta, P.; Rozners, E. Triple-helical recognition of RNA using 2-aminopyridine-modified PNA at physiologically relevant conditions. Angew. Chem. Int. Ed. 2012, 51, 12593–12596. [Google Scholar] [CrossRef] [PubMed]
- Zengeya, T.; Gupta, P.; Rozners, E. Sequence selective recognition of double-stranded RNA using triple helix-forming peptide nucleic acids. Methods Mol. Biol. 2014, 1050, 83–94. [Google Scholar] [PubMed]
- Kim, K.T.; Chang, D.L.; Winssinger, N. Double-stranded RNA-specific templated reaction with triplex forming PNA. Helv. Chim Acta 2018, 101, e1700295. [Google Scholar] [CrossRef]
- Artigas, G.; Marchan, V. Synthesis and tau RNA binding evaluation of ametantrone-containing ligands. J. Org. Chem. 2015, 80, 2155–2164. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ong, A.A.L.; Tan, J.; Bhadra, M.; Dezanet, C.; Patil, K.M.; Chong, M.S.; Kierzek, R.; Decout, J.-L.; Roca, X.; Chen, G. RNA Secondary Structure-Based Design of Antisense Peptide Nucleic Acids for Modulating Disease-Associated Aberrant Tau Pre-mRNA Alternative Splicing. Molecules 2019, 24, 3020. https://doi.org/10.3390/molecules24163020
Ong AAL, Tan J, Bhadra M, Dezanet C, Patil KM, Chong MS, Kierzek R, Decout J-L, Roca X, Chen G. RNA Secondary Structure-Based Design of Antisense Peptide Nucleic Acids for Modulating Disease-Associated Aberrant Tau Pre-mRNA Alternative Splicing. Molecules. 2019; 24(16):3020. https://doi.org/10.3390/molecules24163020
Chicago/Turabian StyleOng, Alan Ann Lerk, Jiazi Tan, Malini Bhadra, Clément Dezanet, Kiran M. Patil, Mei Sian Chong, Ryszard Kierzek, Jean-Luc Decout, Xavier Roca, and Gang Chen. 2019. "RNA Secondary Structure-Based Design of Antisense Peptide Nucleic Acids for Modulating Disease-Associated Aberrant Tau Pre-mRNA Alternative Splicing" Molecules 24, no. 16: 3020. https://doi.org/10.3390/molecules24163020