
Halogen and Hydrogen Bonding in Halogenabenzene/NH3 Complexes Compared Using Next-Generation QTAIM

 ,
,  ,
, 
Abstract
:
1. Introduction
2. Theoretical Background
3. Computational Details
4. Results and Discussion
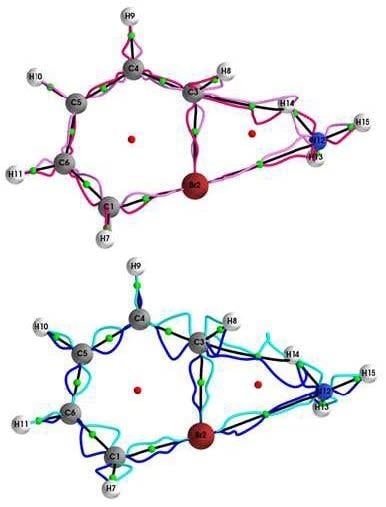
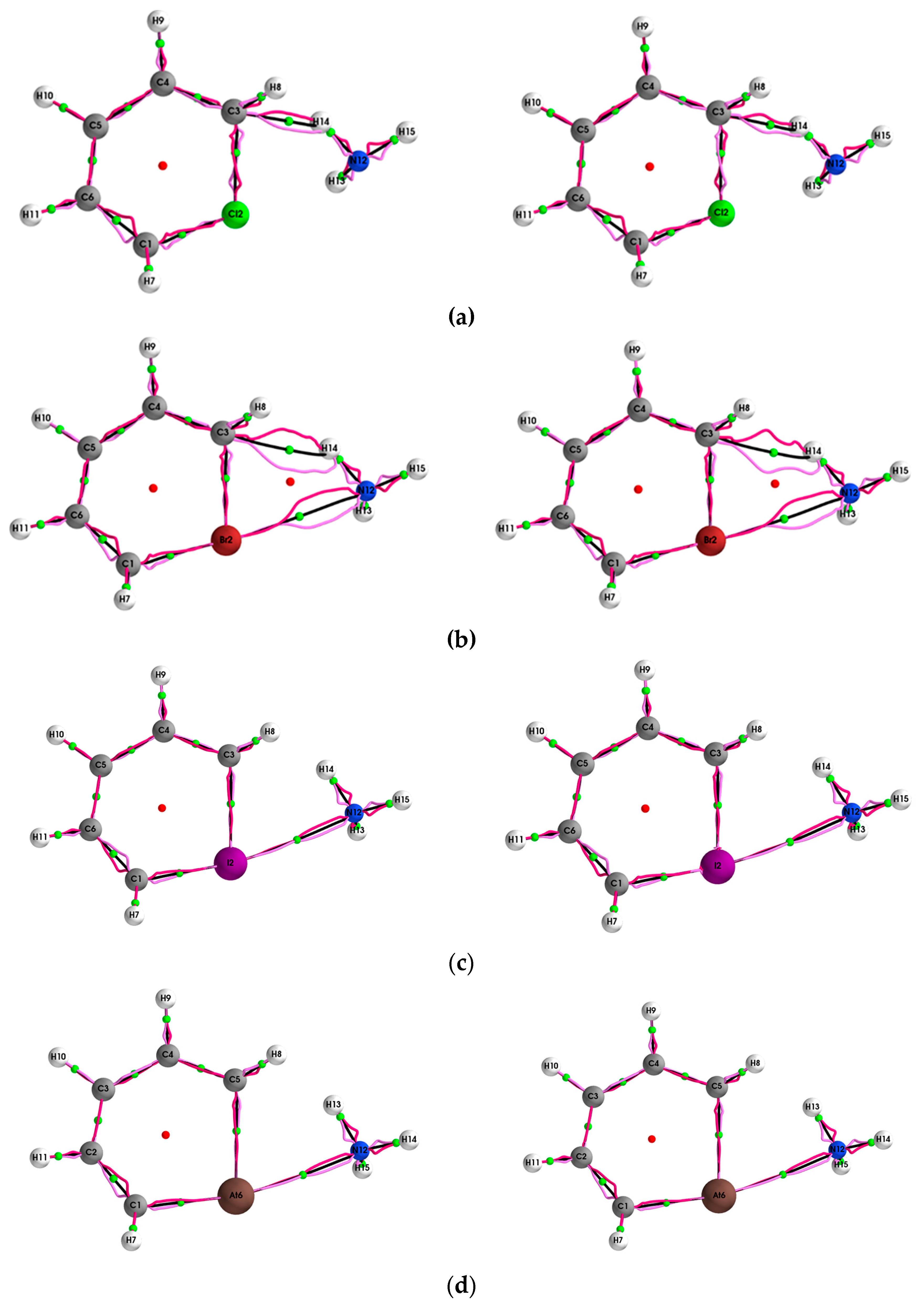
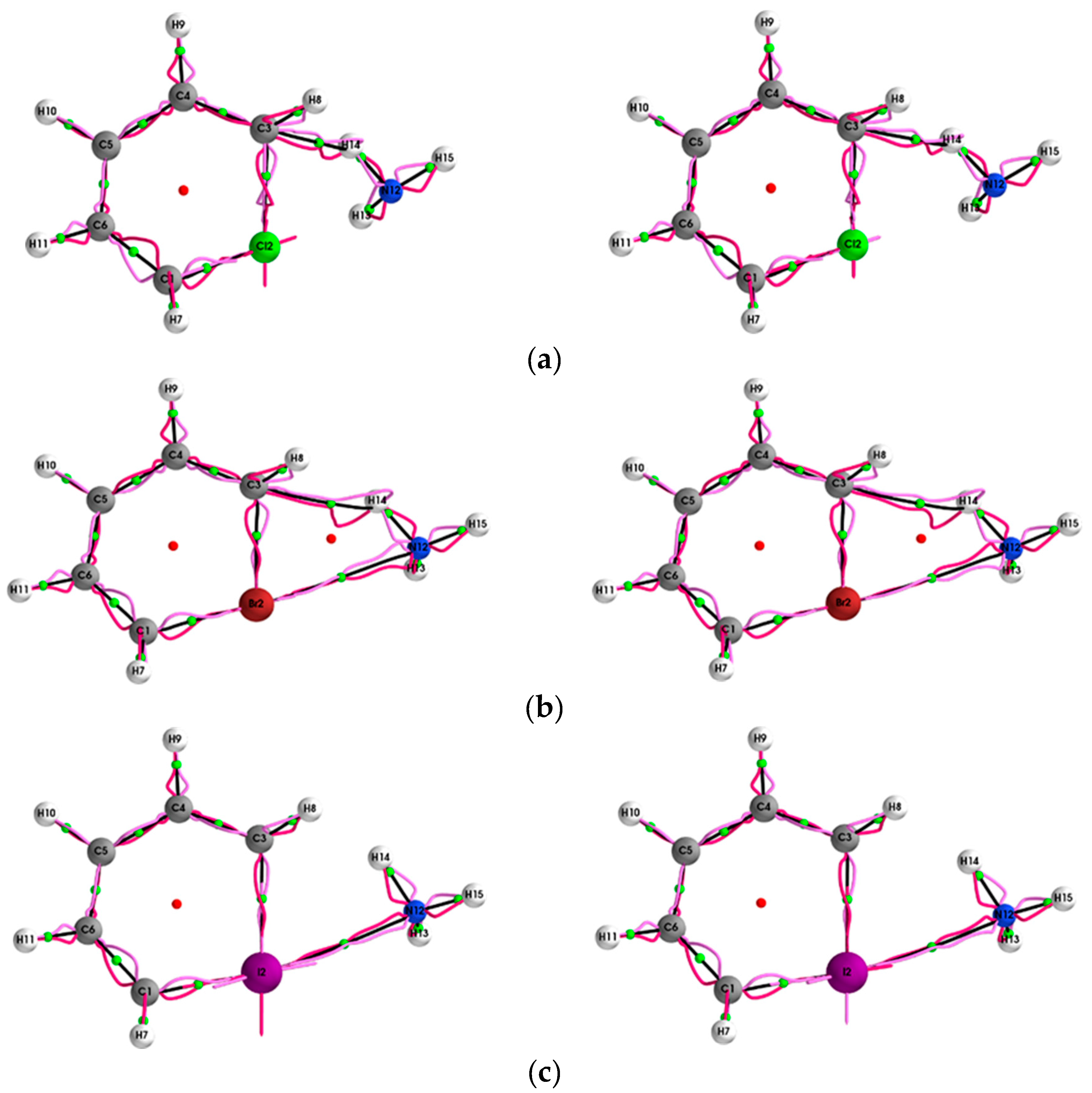
4.1. The QTAIM and Stress Tensor BCP and Bond-Path Properties of the (Y = Cl, Br, I, At)/NH3 System
4.2. Comparison of SR-ZORA and ECPs for QTAIM and Stress Tensor Properties of the (Y = Cl, Br, I, At)/NH3 System
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.; Murray, J.S. An overview of halogen bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Hogan, S.W.L.; van Mourik, T. Competition between hydrogen and halogen bonding in halogenated 1-methyluracil: Water systems. J. Comput. Chem. 2016, 37, 763–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cates, E.L.; van Mourik, T. Halogen bonding with the halogenabenzene bird structure, halobenzene, and halocyclopentadiene. J. Comput. Chem. 2019, 40, 2111–2118. [Google Scholar] [CrossRef] [PubMed]
- Rawashdeh, A.M.; Chakkingal Parambil, P.; Zeng, T.; Hoffmann, R. An Iodabenzene Story. J. Am. Chem. Soc. 2017, 139, 7124–7129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huan, G.; Xu, T.; Momen, R.; Wang, L.; Ping, Y.; Kirk, S.R.; Jenkins, S.; van Mourik, T. A QTAIM exploration of the competition between hydrogen and halogen bonding in halogenated 1-methyluracil: Water systems. Chem. Phys. Lett. 2016, 662, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Pike, S.J.; Hunter, C.A.; Brammer, L.; Perutz, R.N. Benchmarking of Halogen Bond Strength in Solution with Nickel Fluorides: Bromine versus Iodine and Perfluoroaryl versus Perfluoroalkyl Donors. Chem.–Eur. J. 2019, 25, 9237–9241. [Google Scholar] [CrossRef] [PubMed]
- Poster List. Faraday Discuss. 2017, 203, 509–510. [CrossRef]
- Guevara-Vela, J.M.; Ochoa-Resendiz, D.; Costales, A.; Hernández-Lamoneda, R.; Martín Pendás, Á. Halogen Bonds in Clathrate Cages: A Real Space Perspective. ChemPhysChem 2018, 19, 2512–2517. [Google Scholar] [CrossRef] [Green Version]
- Legon, A.C. The halogen bond: An interim perspective. Phys. Chem. Chem. Phys. 2010, 12, 7736–7747. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Halogen Bonding: An Interim Discussion. ChemPhysChem 2013, 14, 278–294. [Google Scholar] [CrossRef]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef] [PubMed]
- Wolters, L.P.; Schyman, P.; Pavan, M.J.; Jorgensen, W.L.; Bickelhaupt, F.M.; Kozuch, S. The many faces of halogen bonding: A review of theoretical models and methods. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 523–540. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolář, M.H.; Hobza, P. Computer Modeling of Halogen Bonds and Other σ-Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [PubMed]
- Kozuch, S.; Martin, J.M.L. Halogen Bonds: Benchmarks and Theoretical Analysis. J. Chem. Theory Comput. 2013, 9, 1918–1931. [Google Scholar] [PubMed]
- Baerends, E.J. Relativistic effects in the electronic-structure and bonding of heavy-metal (u, hg, au) compounds. Abstr. Pap. Am. Chem. Soc. 1984, 187, 89–INOR. [Google Scholar]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1994. [Google Scholar]
- Wang, L.; Ping, Y.; Momen, R.; Azizi, A.; Xu, T.; Rodríguez, J.I.; Anderson, J.S.M.; Kirk, S.R.; Jenkins, S. Insights into the all-metal [Sb3Au3Sb3]3− sandwich complex from a QTAIM and stress tensor analysis. Chem. Phys. Lett. 2017, 685, 127–132. [Google Scholar] [CrossRef]
- Xu, T.; Li, J.H.; Momen, R.; Huang, W.J.; Kirk, S.R.; Shigeta, Y.; Jenkins, S. Chirality-Helicity Equivalence in the S and R Stereoisomers: A Theoretical Insight. J. Am. Chem. Soc. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, S.; Maza, J.R.; Xu, T.; Jiajun, D.; Kirk, S.R. Biphenyl: A stress tensor and vector-based perspective explored within the quantum theory of atoms in molecules. Int. J. Quantum Chem. 2015, 115, 1678–1690. [Google Scholar] [CrossRef]
- Kraka, E.; Cremer, D. Description of chemical reactions in terms of the properties of the electron density. J. Mol. Struct. THEOCHEM 1992, 255, 189–206. [Google Scholar] [CrossRef]
- Jenkins, S.; Blancafort, L.; Kirk, S.R.; Bearpark, M.J. The response of the electronic structure to electronic excitation and double bond torsion in fulvene: A combined QTAIM, stress tensor and MO perspective. Phys. Chem. Chem. Phys. 2014, 16, 7115–7126. [Google Scholar] [CrossRef]
- Jenkins, S. Direct space representation of metallicity and structural stability in SiO solids. J. Phys. Condens. Matter 2002, 14, 10251–10263. [Google Scholar] [CrossRef]
- Jenkins, S.; Ayers, P.W.; Kirk, S.R.; Mori-Sánchez, P.; Martín Pendás, A. Bond metallicity of materials from real space charge density distributions. Chem. Phys. Lett. 2009, 471, 174–177. [Google Scholar] [CrossRef]
- Ayers, P.W.; Jenkins, S. Bond metallicity measures. Comput. Theor. Chem. 2015, 1053, 112–122. [Google Scholar] [CrossRef]
- Jenkins, S.; Heggie, M.I. Quantitative analysis of bonding in 90° partial dislocation in diamond. J. Phys. Condens. Matter 2000, 12, 10325. [Google Scholar] [CrossRef]
- Jenkins, S.; Morrison, I. The chemical character of the intermolecular bonds of seven phases of ice as revealed by ab initio calculation of electron densities. Chem. Phys. Lett. 2000, 317, 97–102. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Nguyen-Dang, T.T. Quantum Theory of Atoms in Molecules–Dalton Revisited. Adv. Quantum Chem. 1981, 14, 63–124. [Google Scholar]
- Bader, R.F.W. Quantum topology of molecular charge distributions. III. The mechanics of an atom in a molecule. J. Chem. Phys. 1980, 73, 2871–2883. [Google Scholar] [CrossRef]
- Guevara-García, A.; Echegaray, E.; Toro-Labbe, A.; Jenkins, S.; Kirk, S.R.; Ayers, P.W. Pointing the way to the products? Comparison of the stress tensor and the second-derivative tensor of the electron density. J. Chem. Phys. 2011, 134, 234106. [Google Scholar] [CrossRef]
- Ayers, P.W.; Jenkins, S. An electron-preceding perspective on the deformation of materials. J. Chem. Phys. 2009, 130, 154104. [Google Scholar] [CrossRef]
- Guevara-García, A.; Ayers, P.W.; Jenkins, S.; Kirk, S.R.; Echegaray, E.; Toro-Labbe, A. Electronic Stress as a Guiding Force for Chemical Bonding. In Electronic Effects in Organic Chemistry; Kirchner, B., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 103–124. [Google Scholar]
- Schwabe, T.; Grimme, S. Towards chemical accuracy for the thermodynamics of large molecules: New hybrid density functionals including non-local correlation effects. Phys. Chem. Chem. Phys. 2006, 8, 4398–4401. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Peterson, K.A.; Figgen, D.; Goll, E.; Stoll, H.; Dolg, M. Systematically convergent basis sets with relativistic pseudopotentials. II. Small-core pseudopotentials and correlation consistent basis sets for the post-d group 16–18 elements. J. Chem. Phys. 2003, 119, 11113–11123. [Google Scholar] [CrossRef]
- Peterson, K.A.; Shepler, B.C.; Figgen, D.; Stoll, H. On the Spectroscopic and Thermochemical Properties of ClO, BrO, IO, and Their Anions. J. Phys. Chem. A 2006, 110, 13877–13883. [Google Scholar] [CrossRef]
- Goerigk, L.; Mehta, N. A Trip to the Density Functional Theory Zoo: Warnings and Recommendations for the User*. Aust. J. Chem. 2019. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, G.; Scuseria, M.; Robb, J.; Cheeseman, J.; Montgomery, T.; Vreven, K.; Kudin, J.; Burant, J.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Zheng, J.; Xu, X.; Truhlar, D.G. Minimally augmented Karlsruhe basis sets. Theor. Chem. Acc. 2011, 128, 295–305. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Neese, F. All-electron scalar relativistic basis sets for the 6p elements. Theor. Chem. Acc. 2012, 131, 1292. [Google Scholar] [CrossRef]
- Aravena, D.; Neese, F.; Pantazis, D.A. Improved Segmented All-Electron Relativistically Contracted Basis Sets for the Lanthanides. J. Chem. Theory Comput. 2016, 12, 1148–1156. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll, Revision 17.01.25, (TK Gristmill Software, 2017).
- Ramachandran, P.; Varoquaux, G. Mayavi: 3D Visualization of Scientific Data. Comput. Sci. Eng. 2011, 13, 40–51. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |
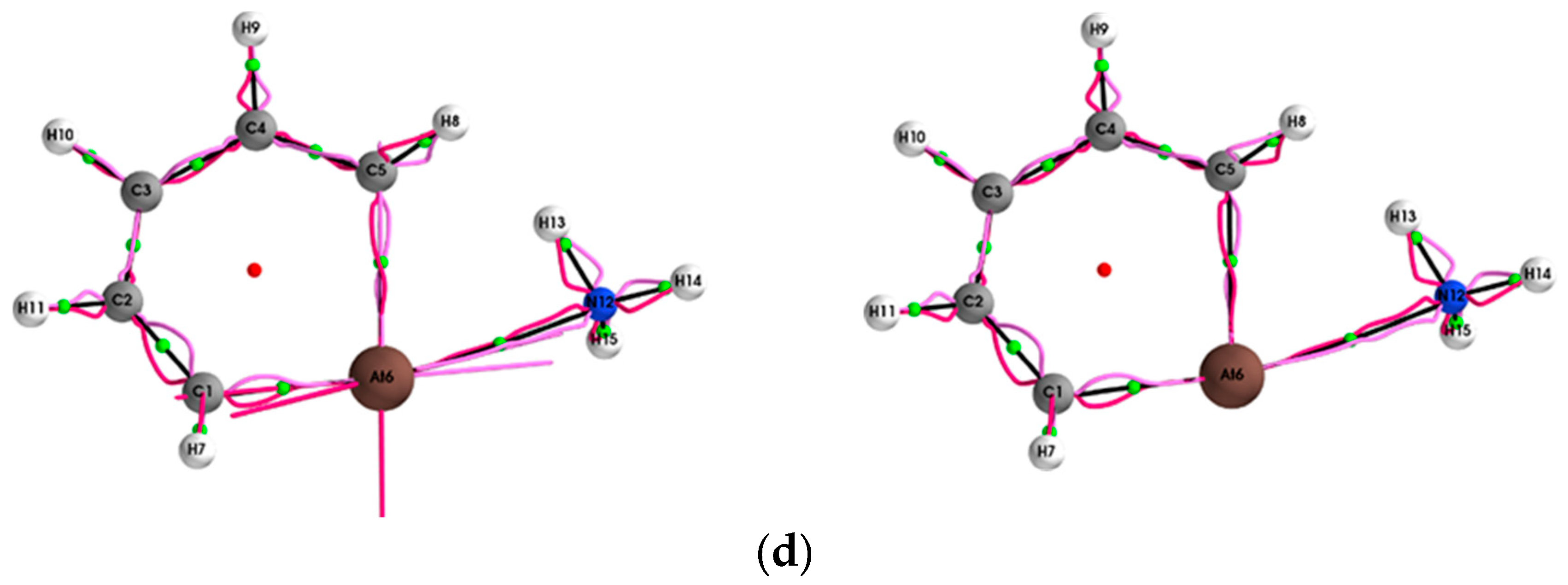
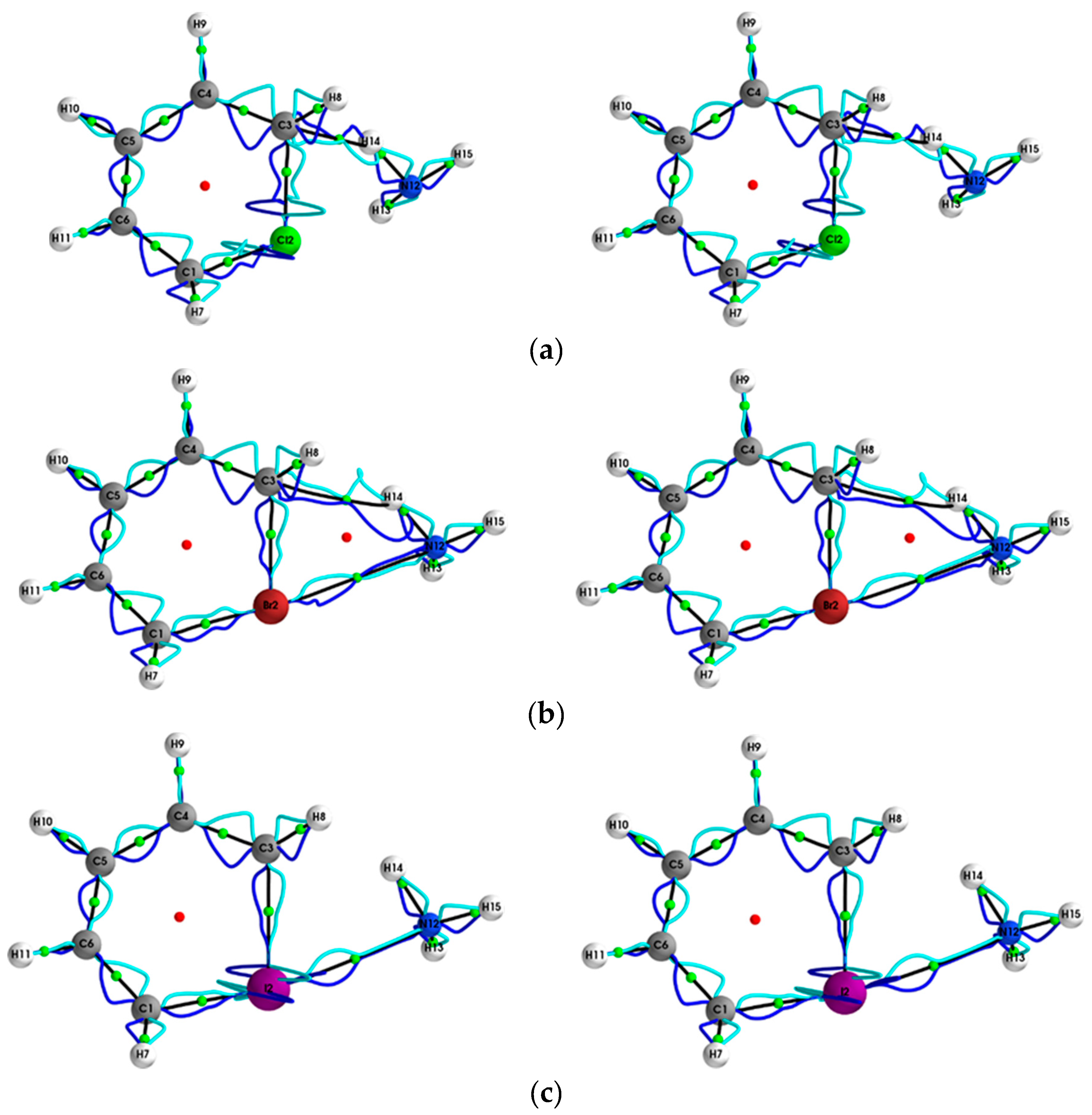


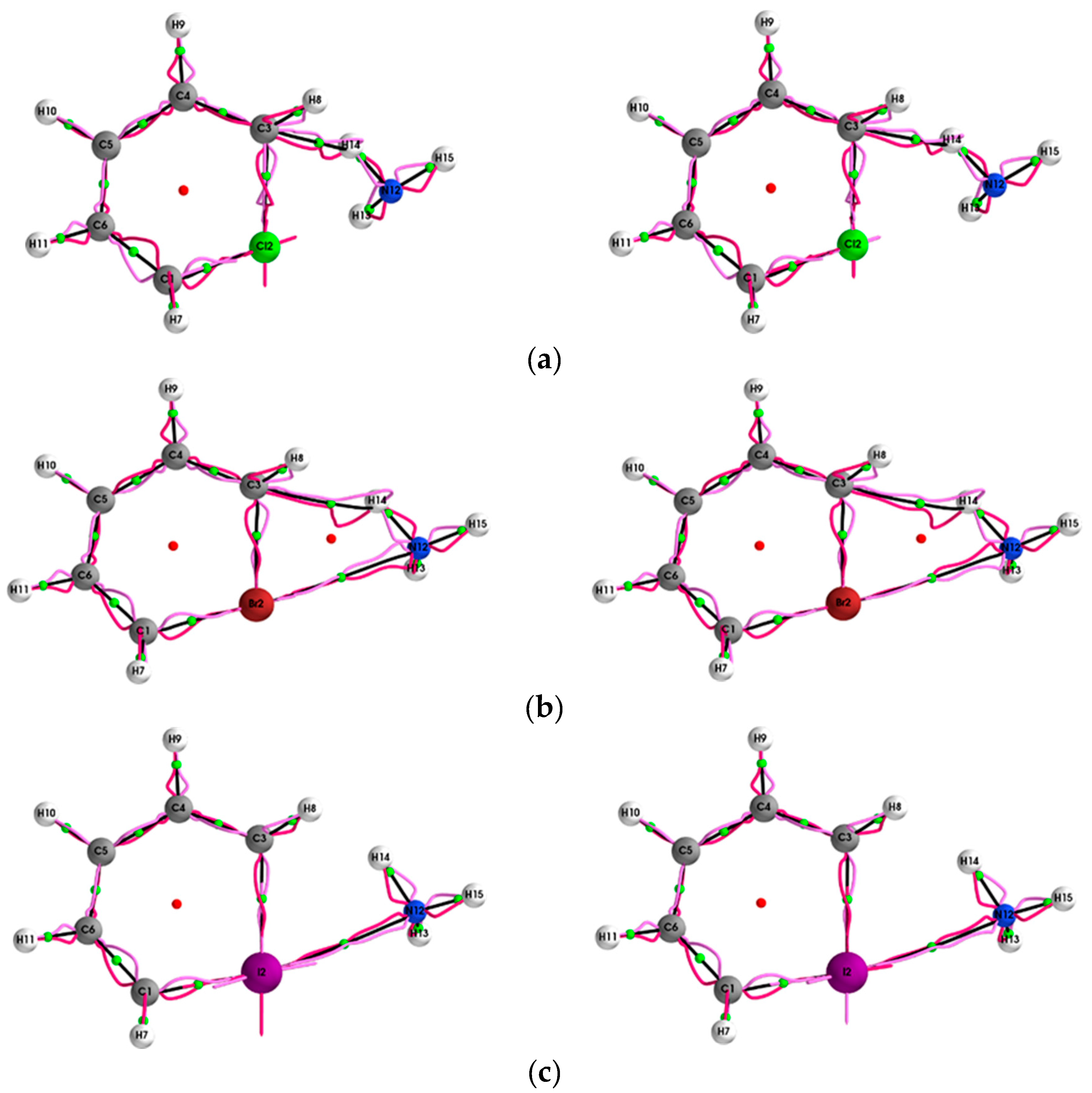
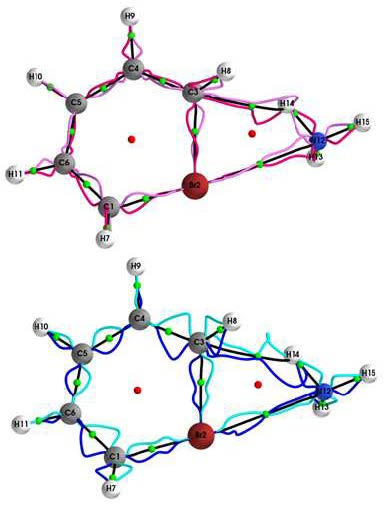{kind=link}
{kind=link}
{kind=link}
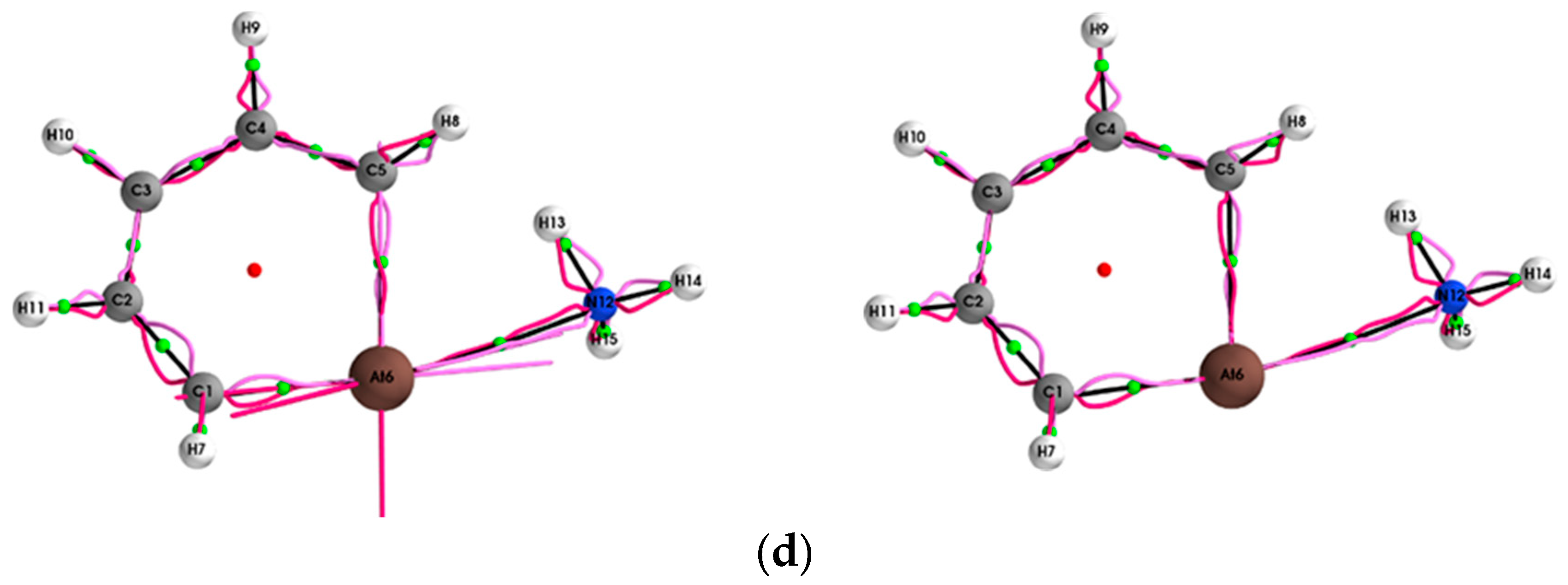{kind=link}
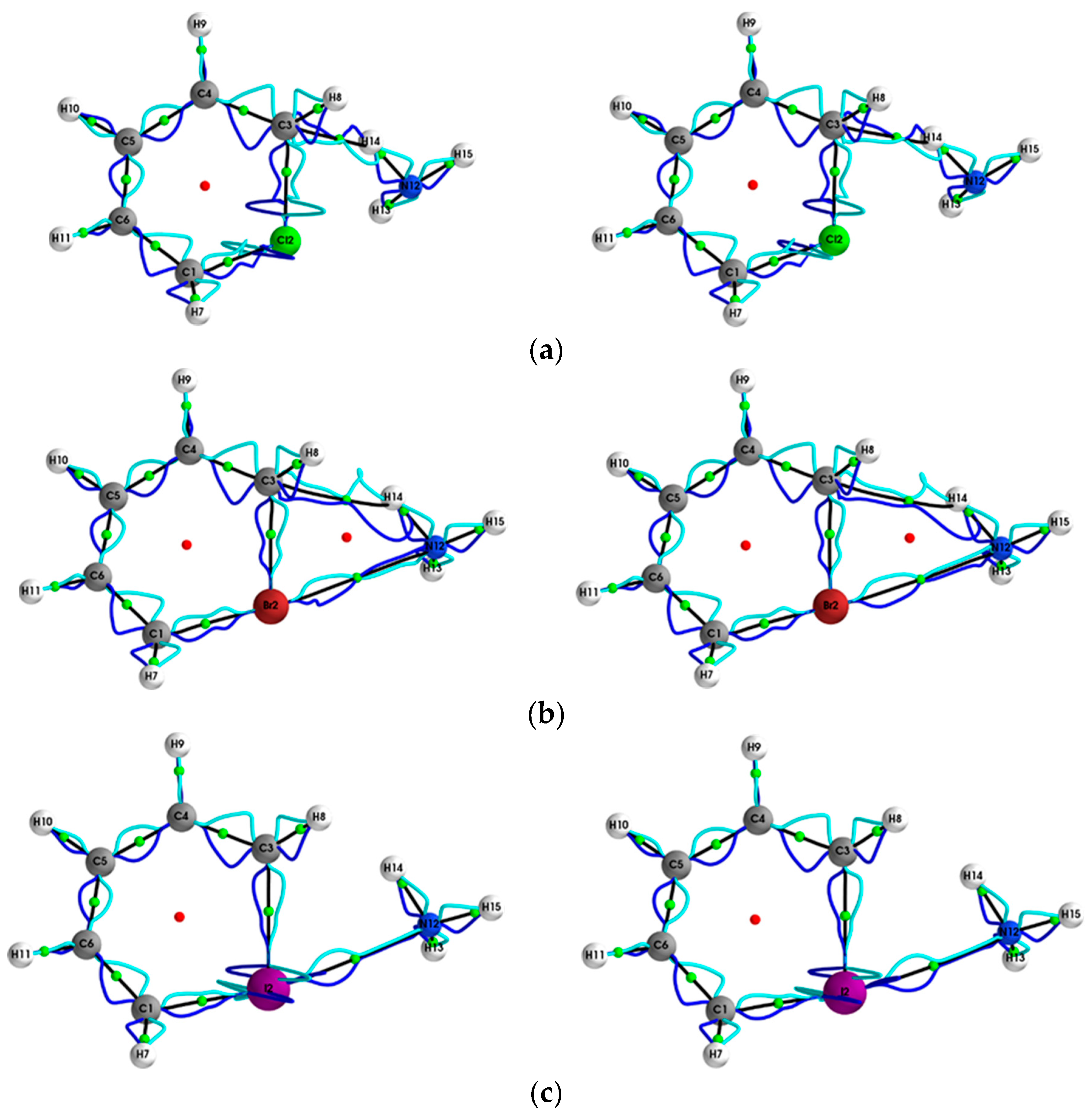{kind=link}
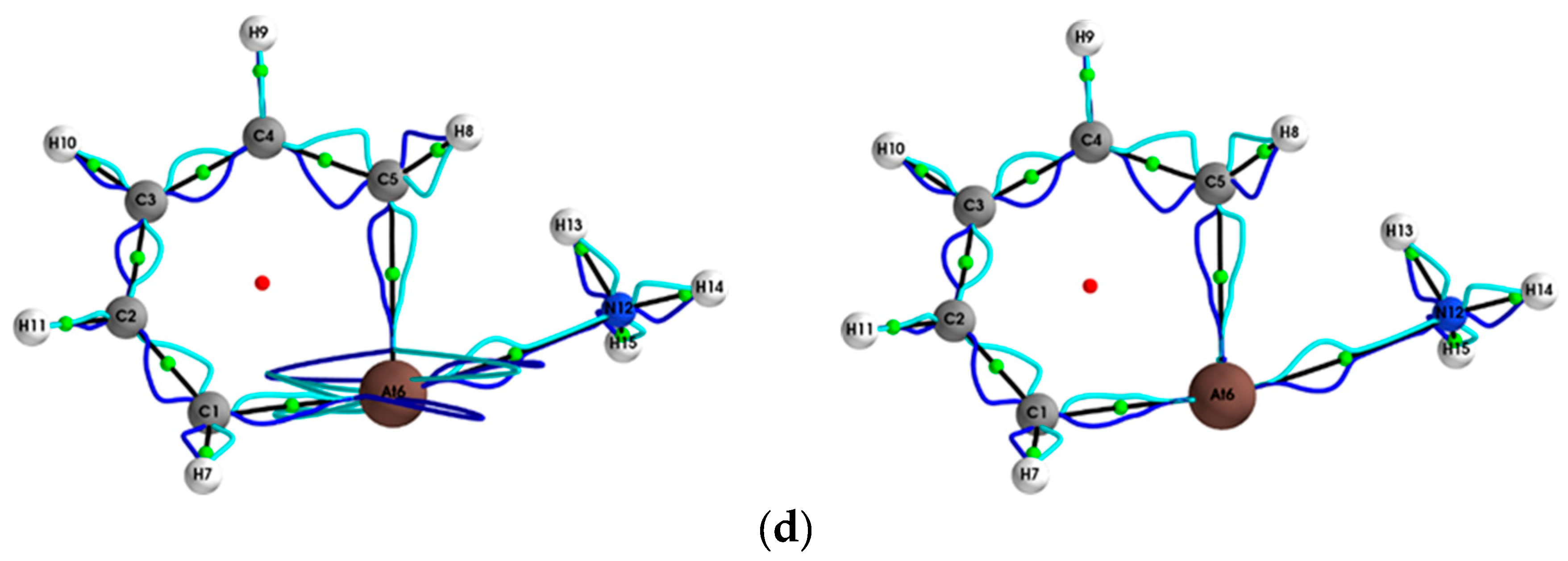{kind=link}
| BCP | (H,H’) | (H*,H*’) | (BPL, GBL) | (Hσ,Hσ’) | (Hσ*,Hσ*’) | ξ(rb) | S | H(rb) |
|---|---|---|---|---|---|---|---|---|
| Cl–NH3 | ||||||||
| C3--H14 | (5.390, 5.202) | (5.266, 5.286) | (4.927, 4.864) | (5.024, 4.928) | (4.968, 4.978) | 0.351 | 0.156 | 0.001 |
| N12-H14 | (2.161, 2.154) | (2.158, 2.158) | (1.877, 1.914) | (1.897, 1.897) | (1.897, 1.897) | −0.198 | 1.496 | −0.481 |
| C3-H8 | (2.317, 2.298) | (2.294, 2.321) | (2.007, 2.036) | (2.051, 2.039) | (2.041, 2.049) | −0.274 | 1.426 | −0.299 |
| C4-C3 | (3.105, 3.095) | (3.082, 3.119) | (2.586, 2.585) | (2.648, 2.631) | (2.637, 2.642) | −0.324 | 1.931 | −0.375 |
| Cl2-C3 | (3.876, 3.831) | (3.839, 3.860) | (3.421. 3.396) | (3.999, 3.983) | (3.859, 3.905) | −0.862 | 0.776 | −0.123 |
| Br–NH3 | ||||||||
| C3--H14 | (6.297, 5.619) | (5.870, 5.905) | (5.125, 4.991) | (5.347, 5.097) | (5.212, 5.223) | 0.329 | 0.115 | 0.001 |
| Br2--N12 | (6.429, 6.376) | (6.358, 6.437) | (6.107, 6.091) | (6.137, 6.133) | (6.129, 6.139) | 0.286 | 0.108 | 0.001 |
| N12-H14 | (2.157, 2.149) | (2.153, 2.153) | (1.876, 1.913) | (1.896, 1.896) | (1.896, 1.896) | −0.198 | 1.500 | −0.482 |
| C3-H8 | (2.294, 2.275) | (2.271, 2.298) | (2.008, 2.037) | (2.051, 2.038) | (2.040, 2.048) | −0.276 | 1.427 | −0.297 |
| C4-C3 | (3.062, 3.054) | (3.038, 3.079) | (2.587, 2.585) | (2.646, 2.627) | (2.634, 2.639) | −0.325 | 1.927 | −0.374 |
| Br2-C3 | (3.895, 3.859) | (3.871, 3.879) | (3.691, 3.666) | (3.716, 3.713) | (3.705, 3.724) | −1.103 | 0.717 | −0.088 |
| I–NH3 | ||||||||
| I2--N12 | (6.213, 6.195) | (6.198, 6.202) | (5.986, 5.970) | (6.588, 6.584) | (6.495, 6.564) | 0.346 | 0.153 | 0.001 |
| C3-H8 | (2.250, 2.231) | (2.229, 2.252) | (2.010, 2.039) | (2.045, 2.035) | (2.036, 2.044) | −0.279 | 1.430 | −0.295 |
| C4-C3 | (3.007, 3.002) | (2.985, 3.026) | (2.589, 2.587) | (2.641, 2.624) | (2.632, 2.634) | −0.329 | 1.905 | −0.369 |
| I2-C3 | (4.363, 4.334) | (4.337, 4.343) | (4.025, 4.004) | (4.640, 4.638) | (4.542, 4.628) | −2.200 | 0.600 | −0.061 |
| At–NH3 | ||||||||
| At6--N12 | (6.160, 6.142) | (6.143, 6.146) | (5.927, 5.914) | (5.948, 5.943) | (5.939, 5.944) | 0.357 | 0.158 | 0.001 |
| C5-H8 | (2.257, 2.238) | (2.237, 2.259) | (2.014, 2.043) | (2.051, 2.041) | (2.042, 2.050) | −0.280 | 1.429 | −0.292 |
| C4-C5 | (2.963, 2.957) | (2.939, 2.982) | (2.593, 2.591) | (2.644, 2.626) | (2.633, 2.636) | −0.329 | 1.910 | −0.367 |
| At6-C5 | (4.636, 4.616) | (4.622, 4.623) | (4.235, 4.208) | (4.272, 4.270) | (4.248, 4.265) | −22.03 | 0.486 | −0.046 |
| BCP | ∆(H,H’) (%) | ∆(H*,H*’) (%) | ∆(BPL, GBL) (%) | ∆(Hσ,Hσ’) (%) | ∆(Hσ*,Hσ*’) (%) | ∆ξ(rb) (%) | ∆S (%) | ∆H(rb) (%) |
|---|---|---|---|---|---|---|---|---|
| Cl–NH3 | ||||||||
| C3--H14 | (−0.242, 0.115) | (−0.171, −0.019) | (−0.102, 0.000) | (−0.259, 0.000) | (−0.202, −0.101) | −3.540 | −0.645 | 0.000 |
| N12-H14 | (0.231, 0.185) | (0.185, 0.185) | (−0.321, 0.000) | (−0.264, −0.264) | (−0.264, −0.264) | −5.319 | 8.502 | 8.031 |
| C3-H8 | (2.030, 1.921) | (1.966, 2.026) | (−0.200, 0.000) | (0.195, 0.049) | (0.098, 0.098) | −11.38 | 18.05 | 12.57 |
| C4-C3 | (2.908, 2.826) | (2.868, 2.895) | (0.039, 0.000) | (0.339, 0.303) | (0.265, 0.339) | −13.68 | 36.40 | 10.29 |
| Cl2-C3 | (3.003, 2.643) | (2.786, 2.746) | (0.175, 0.000) | (1.502, 1.872) | (1.656, 1.563) | −15.70 | 9.240 | 6.107 |
| Br–NH3 | ||||||||
| C3--H14 | (−0.671, −0.807) | (−1.224, −0.854) | (−0.039, 0.000) | (−0.300, 0.157) | (−0.192, −0.173) | −1.543 | −2.679 | 0.000 |
| Br2--N12 | (−0.047, 0.530) | (0.235, 0.294) | (0.000, 0.000) | (0.114, −0.114) | (−0.147, 0.065) | −4.380 | −5.882 | 50.00 |
| N12-H14 | (0.416, 0.371) | (0.370, 0.416) | (−0.267, 0.000) | (−0.264, −0.264) | (−0.264, −0.264) | −5.319 | 8.369 | 8.015 |
| C3-H8 | (1.587, 1.515) | (1.518, 1.627) | (−0.150, 0.000) | (0.097, 0.000) | (0.049, 0.049) | −11.74 | 18.32 | 12.90 |
| C4-C3 | (2.608, 2.553) | (2.597, 2.594) | (0.000, 0.000) | (0.264, 0.266) | (0.265, 0.265) | −13.64 | 36.34 | 10.31 |
| Br2-C3 | (2.209, 1.906) | (2.050, 1.946) | (0.135, 0.000) | (0.215, 0.242) | (0.135, 0.321) | −7.926 | 2.846 | 1.124 |
| I–NH3 | ||||||||
| I2--N12 | (−2.339, −2.295) | (−2.243, −2.276) | (−0.050, 0.000) | (0.768, 0.813) | (−0.480, 1.515) | −2.065 | −2.000 | 0.000 |
| C3-H8 | (−0.852, −0.768) | (−0.814, −0.806) | (−0.100, 0.000) | (−0.196, −0.197) | (−0.197, −0.245) | −10.28 | 15.48 | 11.68 |
| C4-C3 | (−1.008, −0.975) | (−0.981, −1.035) | (0.000, 0.000) | (−0.152, −0.114) | (−0.152, −0.152) | −10.77 | 28.89 | 8.889 |
| I2-C3 | (−4.880, −4.965) | (−4.683, −4.827) | (−0.025, 0.000) | (1.003, 1.003) | (−0.643, 1.845) | −19.24 | 3.692 | 4.688 |
| At–NH3 | ||||||||
| At6--N12 | (−2.564, −2.555) | (−2.452, −2.467) | (−0.034, 0.000) | (18.07, 18.13) | (12.62, 19.96) | −4.386 | −1.282 | 0.000 |
| C5-H8 | (−0.267, −0.314) | (−0.314, −0.311) | (−0.099, 0.000) | (−0.146, −0.196) | (−0.196, −0.196) | −10.24 | 15.29 | 11.78 |
| C4-C5 | (−0.407, −0.373) | (−0.376, −0.404) | (0.000, 0.000) | (−0.114, −0.114) | (−0.076, −0.114) | −10.40 | 27.92 | 8.706 |
| At6-C5 | (−7.042, −7.174) | (−7.040, −7.039) | (0.000, 0.000) | (23.23, 23.24) | (17.11, 25.71) | 1.432 | −0.621 | 2.128 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Xu, T.; van Mourik, T.; Früchtl, H.; Kirk, S.R.; Jenkins, S. Halogen and Hydrogen Bonding in Halogenabenzene/NH3 Complexes Compared Using Next-Generation QTAIM. Molecules 2019, 24, 2875. https://doi.org/10.3390/molecules24162875
Li S, Xu T, van Mourik T, Früchtl H, Kirk SR, Jenkins S. Halogen and Hydrogen Bonding in Halogenabenzene/NH3 Complexes Compared Using Next-Generation QTAIM. Molecules. 2019; 24(16):2875. https://doi.org/10.3390/molecules24162875
Chicago/Turabian StyleLi, Shuman, Tianlv Xu, Tanja van Mourik, Herbert Früchtl, Steven R. Kirk, and Samantha Jenkins. 2019. "Halogen and Hydrogen Bonding in Halogenabenzene/NH3 Complexes Compared Using Next-Generation QTAIM" Molecules 24, no. 16: 2875. https://doi.org/10.3390/molecules24162875
APA StyleLi, S., Xu, T., van Mourik, T., Früchtl, H., Kirk, S. R., & Jenkins, S. (2019). Halogen and Hydrogen Bonding in Halogenabenzene/NH3 Complexes Compared Using Next-Generation QTAIM. Molecules, 24(16), 2875. https://doi.org/10.3390/molecules24162875