Lignan Glycosides from Urena lobata



Abstract
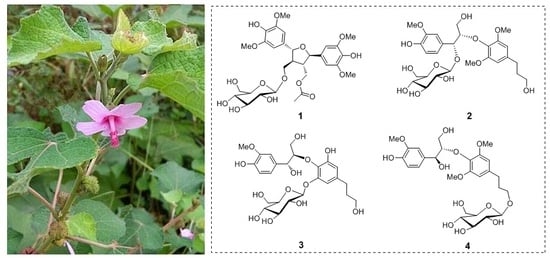
:
1. Introduction
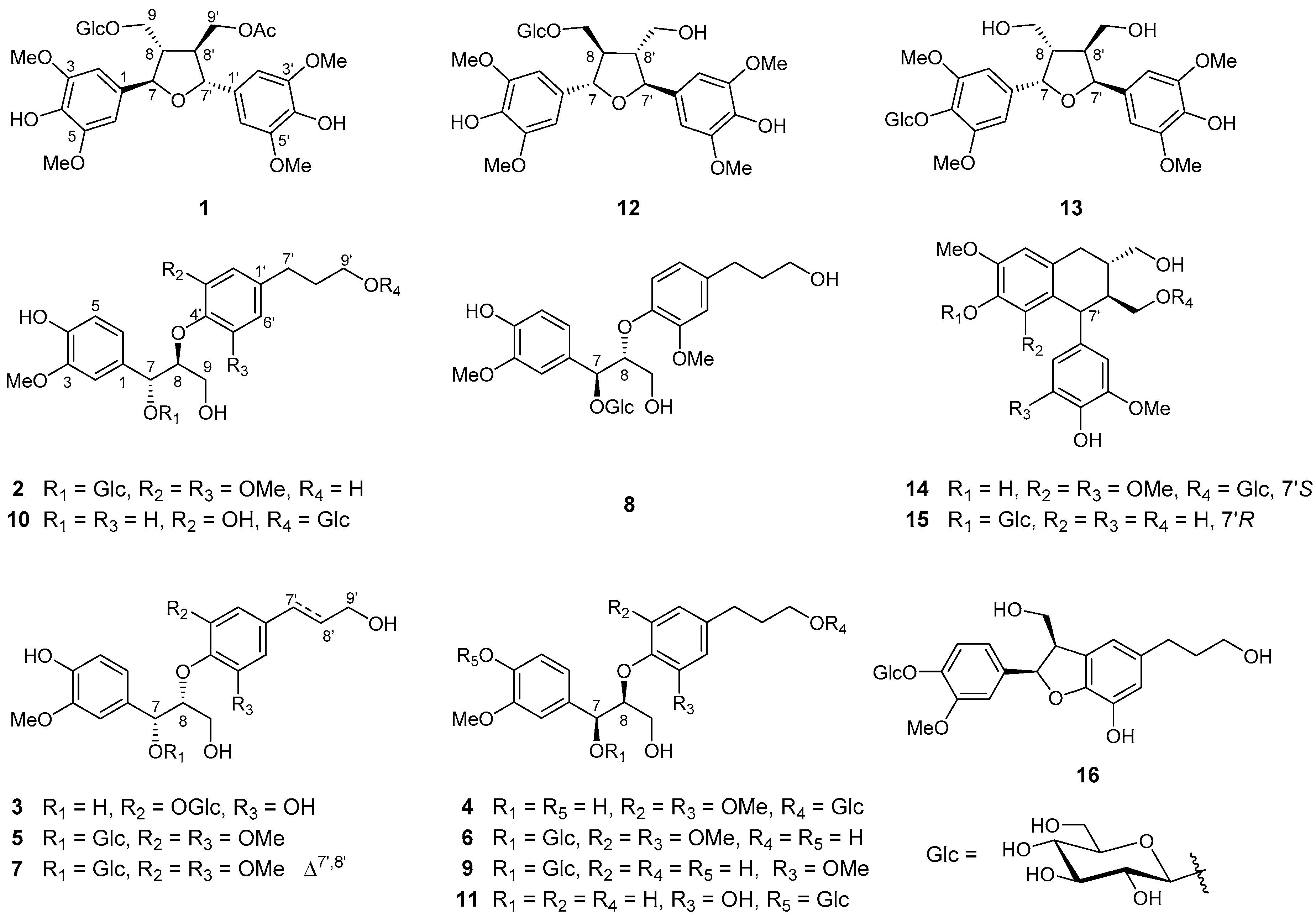
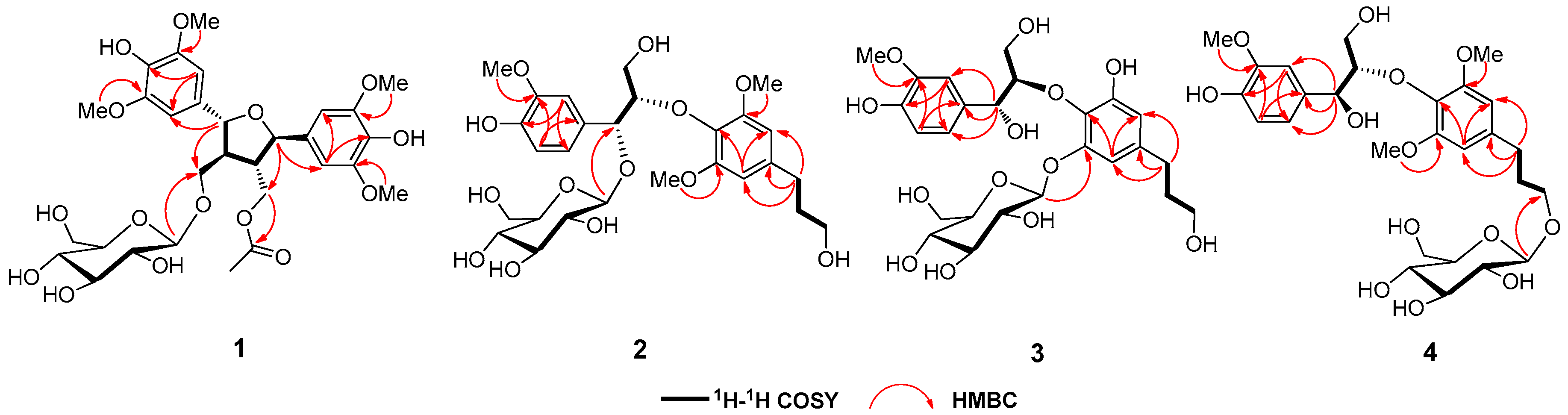
2. Results
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Biological Assays
3.5. Bioactivity Evaluation
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Babu, S.S.; Madhuri, D.B.; Ali, S.L. A pharmacological review of Urena lobata plant. Asian. J. Pharm. Clin. Res. 2016, 9, 20–22. [Google Scholar]
- Singh, D.; Singh, V. Isolation and characterization of flavonoids in Urena lobata leaves. J. Neurosci NLM. 2016, 20, 845–853. [Google Scholar] [CrossRef]
- Chinese Pharmacopoeia Commission. Pharmacopoeia of the People’s Republic of China, 9th ed.; China Medical Science and Technology Press: Beijing, China, 2010; Volume 1, p. 97. [Google Scholar]
- Sajem, A.L.; Gosai, K. Traditional use of medicinal plants by the Jaintia tribes in North Cachar Hills district of Assam, northeast India. J. Ethnobiol. Ethnomed. 2006, 2, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, U.K.; Gupta, M.; Manikandan, L.; Bhattacharya, S. Antibacterial activity of Urena lobata root. Fitoterapia 2001, 72, 927–929. [Google Scholar] [CrossRef]
- Heras, B.D.L.; Slowing, K.; Benedí, J.; Carretero, E.; Ortege, T.; Toledo, C.; Bermejo, P.; Iglesias, I.; Abad, M.J.; Gomez-Serranillos, P.; et al. Antiinflammatory and antioxidant activity of plants used in traditional medicine in Ecuador. J. Ethnopharmacol. 1998, 61, 161–166. [Google Scholar] [CrossRef]
- Pieme, C.A.; Penlap, V.N.; Ngogang, J.; Costache, M. In vitro cytotoxicity and antioxidant activities of five medicinal plants of Malvaceae family from Cameroon. Environ. Toxicol. Pharmacol. 2010, 29, 220–228. [Google Scholar] [CrossRef] [PubMed]
- An Editorial Committee of the Administration Bureau of Traditional Chinese Medicine. Chinese Materia Medica (Zhonghua Bencao), 1st ed.; Shanghai Science &Technology Press: Shanghai, China, 1999; Volume 14, pp. 372–373. [Google Scholar]
- Su, C.; Yang, W.Q.; Jiang, D.; Zhang, X.; Zheng, J.; Shi, S.P.; Tu, P.F. Flavonoids from Urena lobata. Chin. Tradit. Herbal. Drugs 2015, 46, 2034–2039. [Google Scholar]
- Shi, X.P.; Su, C.; Qi, B.W.; Yang, W.Q.; Wu, Y.; Ding, N.; Dong, X.J.; Shi, S.P. Flavonoid glycosides from Aerial part of Urena lobate. Chin. Pharm. J. 2017, 52, 19–23. [Google Scholar]
- Su, C.; Yang, W.Q.; Li, B.; Zhang, X.; Gao, B.W.; Tu, P.F.; Shi, S.P. Quantification of tiliroside in Urena lobata L. by HPLC. Chin. New. Drug. J. 2015, 24, 2865–2867. [Google Scholar]
- Gao, X.; Liao, Y.; Wang, J.; Liu, X.; Zhong, K.; Huang, Y. Discovery of a potent anti-yeast triterpenoid saponin, clematoside-S from Urena lobata L. Int. J. Mol. Sci. 2015, 16, 4731–4743. [Google Scholar] [CrossRef]
- Morelli, G.F.; Cairoli, P.; Speranza, G.; Alamgir, M.; Rajia, S. Triglycerides from Urena lobata. Fitoterapia 2006, 77, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Qi, B.; Wang, J.; Ding, N.; Wu, Y.; Shi, X.P.; Zhu, Z.X.; Liu, X.; Wang, X.H.; Zheng, J.; et al. Megastigmane glycosides from Urena lobata. Fitoterapia 2018, 127, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, H.; Miyase, T.; Ueno, A. Lignan and terpene glycosides from Epimedium sagittatum. Phytochemistry 1991, 30, 2025–2027. [Google Scholar] [CrossRef]
- Yamauchi, H.; Kakuda, R.; Yaoita, Y.; Machida, K.; Kikuchi, M. Two new glycosides from the whole plants of Glechoma hederacea L. Chem. Pharm. Bull. 2010, 38, 346–347. [Google Scholar]
- Sugiyama, M.; Nagayama, E.; Kikuchi, M. Lignan and phenylpropanoid glycosides from Osmanthus Asiaticus. Phytochemistry 1993, 33, 1215–1219. [Google Scholar] [CrossRef]
- Jutiviboonsuk, A.; Zhang, H.; Tan, G.T.; Ma, C.; Hung, N.J.; Cuong, M.N.; Bunyapraphatsara, N.; Soejarto, D.D.; Fong, H.H.S. Bioactive constituents from roots of Bursera tonkinensis. Phytochemistry 2005, 66, 2745–2751. [Google Scholar] [CrossRef] [PubMed]
- Besombes, S.; Robert, D.; Utille, J.P.; Taravel, F.R.; Mazeau, K. Molecular modeling of syringyl and p-hydroxyphenyl β-O-4 dimers. Comparative study of the computed and experimental conformational properties of lignin β-O-4 model compounds. J. Agr. Food Chem. 2003, 51, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Braga, A.C.H.; Zacchino, S.; Badano, H.; Sierra, M.G.; Rúveda, E.A. 13C-NMR spectral and conformational analysis of 8-O-4′ neolignans. Phytochemistry 1984, 23, 2025–2028. [Google Scholar] [CrossRef]
- Qu, L.; Ruan, J.Y.; Jin, L.J.; Shi, W.Z.; Li, X.X.; Han, L.F.; Zhang, Y.; Wang, T. Xanthine oxidase inhibitory effects of the constituents of Chrysanthemum morifolium stems. Phytochem. Lett. 2017, 19, 39–45. [Google Scholar] [CrossRef]
- He, Z.D.; Ma, C.Y.; Tan, G.T.; Sydara, K.; Tamez, P.; Southavong, B.; Bouamanivong, S.; Soejarto, D.D.; Pezzuto, J.M.; Fong, H.H.S.; et al. Rourinoside and rouremin, antimalarial constituents from Rourea minor. Phytochemistry 2006, 67, 1378–1384. [Google Scholar] [CrossRef]
- Machida, K.; Sakamoto, S.; Kikuchi, M. Two new neolignan glycosides from leaves of Osmanthus heterophyllus. J. Nat. Med. 2009, 63, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Noriko, M.; Masao, K. Studies on the constituents of Lonicera Species. X. Chem. Pharm. Bull. 1996, 44, 1676–1679. [Google Scholar]
- Gan, M.; Zhang, Y.; Lin, S.; Liu, M.; Song, W.; Zi, J.; Yang, Y.; Fan, X.; Shi, J.; Hu, J.; et al. Glycosides from the root of Iodes cirrhosa. J. Nat. Prod. 2008, 71, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, Z.; Feng, Z.; Jiang, J.; Zhang, P. Lignans from the root of Rhodiola crenulata. J. Agr. Food Chem. 2012, 60, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Góngora, L.; Máñez, S.; Giner, R.M.; Recio, M.C.; Gray, A.I.; Rios, J.L. Phenolic glycosides from Phagnalon rupestre. Phytochemistry 2002, 59, 857–860. [Google Scholar] [CrossRef]
- Ahmad, M.; Akhtar, M.F.; Miyase, T.; Ueno, A.; Rashid, S.; Usmanghani, K. Studies on the medicinal herb Ruellia patula. Pharm. Biol. 1993, 31, 121–129. [Google Scholar] [CrossRef]
- Jiang, Z.H.; Tanaka, T.; Sakamoto, M.; Jiang, T.; Kouno, I. Studies on a medicinal parasitic plant: Lignans from the stems of Cynomorium songaricum. Chem. Pharm. Bull. 2001, 49, 1036–1038. [Google Scholar] [CrossRef]
- Lundgren, L.N.; Popoff, T.; Theander, O. Dilignol glycosides from needles of Picea abies. Phytochemistry 1981, 20, 1967–1969. [Google Scholar] [CrossRef]
- Pan, J.Y.; Chen, S.L.; Yang, M.H.; Wu, J.; Sinkkonen, J.; Zou, K. An update on lignans: Natural products and synthesis. Nat. Prod. Rep. 2009, 26, 1251–1292. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1–16 are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
| No. | 1 a | 2 a | 3 a | 4 a | ||||
|---|---|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | δH | δC | |
| 1 | 134.1 | 130.8 | 133.5 | 133.3 | ||||
| 2 | 6.78, s | 105.2 | 7.28, d, (1.5) | 113.3 | 7.01, d, (1.5) | 111.6 | 7.08, d, (1.5) | 112.2 |
| 3 | 149.4 | 148.7 | 149.0 | 148.5 | ||||
| 4 | 136.3 | 147.1 | 147.5 | 147.0 | ||||
| 5 | 149.4 | 6.83, d, (8.0) | 115.5 | 6.77, d, (8.5) | 116.1 | 6.78, d, (8.0) | 115.7 | |
| 6 | 6.78, s | 105.2 | 6.96, dd, (8.0, 1.5) | 122.1 | 6.88, dd, (8.0, 1.5) | 121.1 | 6.94, dd, (8.0, 1.5) | 121.0 |
| 7 | 5.12, d, (8.0) | 85.6 | 5.31, d, (3.0) | 77.7 | 4.97, d, (8.5) | 75.0 | 5.12, d, (7.0) | 74.4 |
| 8 | 2.75, m | 52.7 | 4.23, m | 86.8 | 4.01, m | 89.7 | 4.16, m | 88.3 |
| 9 | 3.80, dd, (10.0, 4.5) 4.09, dd, (10.0, 5.5) | 69.3 | 3.16, m | 61.4 | 3.69, m | 61.3 | 3.62, m | 62.1 |
| 1′ | 133.6 | 135.1 | 140.0 | 133.3 | ||||
| 2′ | 6.80, s | 104.8 | 6.53, s | 106.7 | 6.46, s | 109.3 | 6.59, s | 106.7 |
| 3′ | 149.3 | 154.3 | 152.0 | 153.9 | ||||
| 4′ | 136.4 | 139.9 | 135.3 | 140.1 | ||||
| 5′ | 149.3 | 154.3 | 152.0 | 153.9 | ||||
| 6′ | 6.80, s | 104.8 | 6.53, s | 106.7 | 6.60, s | 112.1 | 6.59, s | 106.7 |
| 7′ | 4.98, d, (9.0) | 84.2 | 2.67, t, (7.5) | 33.4 | 2.56, t, (7.5) | 33.0 | 2.69, t, (7.5) | 33.4 |
| 8′ | 2.45, m, | 51.1 | 1.86, m | 35.4 | 1.80, m | 35.2 | 1.87, m | 35.4 |
| 9′ | 3.72, (dd,12.0, 5.0) 4.36, overlapped | 64.8 | 3.61, t, (6.4) | 62.8 | 3.56, t, (6.5) | 62.2 | 3.81, dd, (11.0, 2.5) 3.93, dd, (11.0, 4.0) | 69.2 |
| Glu-1′′ | 4.36, d, (8.0) | 104.6 | 4.23, d, (7.5) | 101.0 | 4.93, d, (7.5) | 103.0 | 4.32, d, (8.0) | 104.5 |
| Glu-2′′ | 3.26, overlapped | 75.2 | 3.45, overlapped | 75.2 | 3.48, overlapped | 75.1 | 3.21, overlapped | 75.4 |
| Glu-3′′ | 3.40, overlapped | 78.1 | 3.45, overlapped | 77.8 | 3.41, overlapped | 78.0 | 3.25, overlapped | 77.9 |
| Glu-4′′ | 3.33, overlapped | 71.6 | 3.32, overlapped | 71.9 | 3.40, overlapped | 71.4 | 3.26, overlapped | 71.8 |
| Glu-5′′ | 3.36, overlapped | 78.2 | 3.45, overlapped | 78.1 | 3.47, overlapped | 78.3 | 3.28, overlapped | 78.0 |
| Glu-6′′ | 4.32, overlapped 4.36, overlapped | 62.8 | 3.87, overlapped 3.92, overlapped | 62.2 | 3.68, overlapped 3.89, overlapped | 62.5 | 3.68, overlapped 3.89, overlapped | 62.9 |
| COCH3 | 172.8 | |||||||
| COCH3 | 1.95 (3H, s) | 20.7 | ||||||
| 3-OCH3 | 3.93 (3H, s) | 56.9 | 3.89 (3H, s) | 56.4 | 3.86, (3H, s) | 56.4 | 3.89, (3H, s) | 56.5 |
| 5-OCH3 | 3.93 (3H, s) | 56.9 | ||||||
| 3′-OCH3 | 3.93 (3H, s) | 56.9 | 3.74 (3H, s) | 56.4 | 3.89, (3H, s) | 56.6 | ||
| 5′-OCH3 | 3.93 (3H, s) | 56.9 | 3.74 (3H, s) | 56.4 | 3.89, (3H, s) | 56.6 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, Y.; Su, C.; Ding, N.; Qi, B.; Jia, F.; Xu, X.; Liu, X.; Wang, J.; Wang, X.; Tu, P.; et al. Lignan Glycosides from Urena lobata. Molecules 2019, 24, 2850. https://doi.org/10.3390/molecules24152850
Luo Y, Su C, Ding N, Qi B, Jia F, Xu X, Liu X, Wang J, Wang X, Tu P, et al. Lignan Glycosides from Urena lobata. Molecules. 2019; 24(15):2850. https://doi.org/10.3390/molecules24152850
Chicago/Turabian StyleLuo, Yuan, Cong Su, Ning Ding, Bowen Qi, Fangfang Jia, Xiping Xu, Xiao Liu, Juan Wang, Xiaohui Wang, Pengfei Tu, and et al. 2019. "Lignan Glycosides from Urena lobata" Molecules 24, no. 15: 2850. https://doi.org/10.3390/molecules24152850
APA StyleLuo, Y., Su, C., Ding, N., Qi, B., Jia, F., Xu, X., Liu, X., Wang, J., Wang, X., Tu, P., & Shi, S. (2019). Lignan Glycosides from Urena lobata. Molecules, 24(15), 2850. https://doi.org/10.3390/molecules24152850