
Analysis of the Carry-Over of Ochratoxin A from Feed to Milk, Blood, Urine, and Different Tissues of Dairy Cows Based on the Establishment of a Reliable LC-MS/MS Method


Abstract
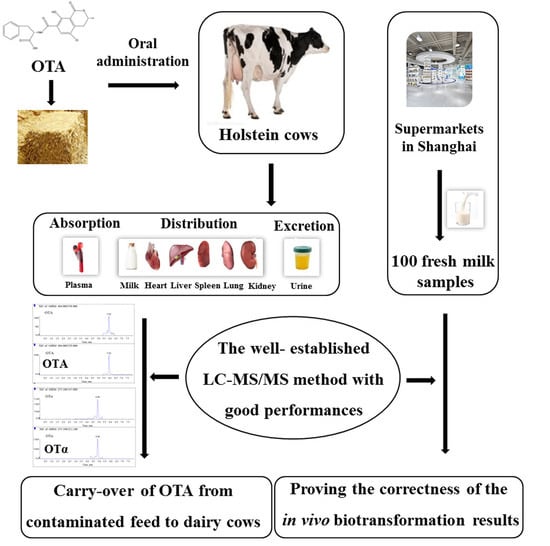
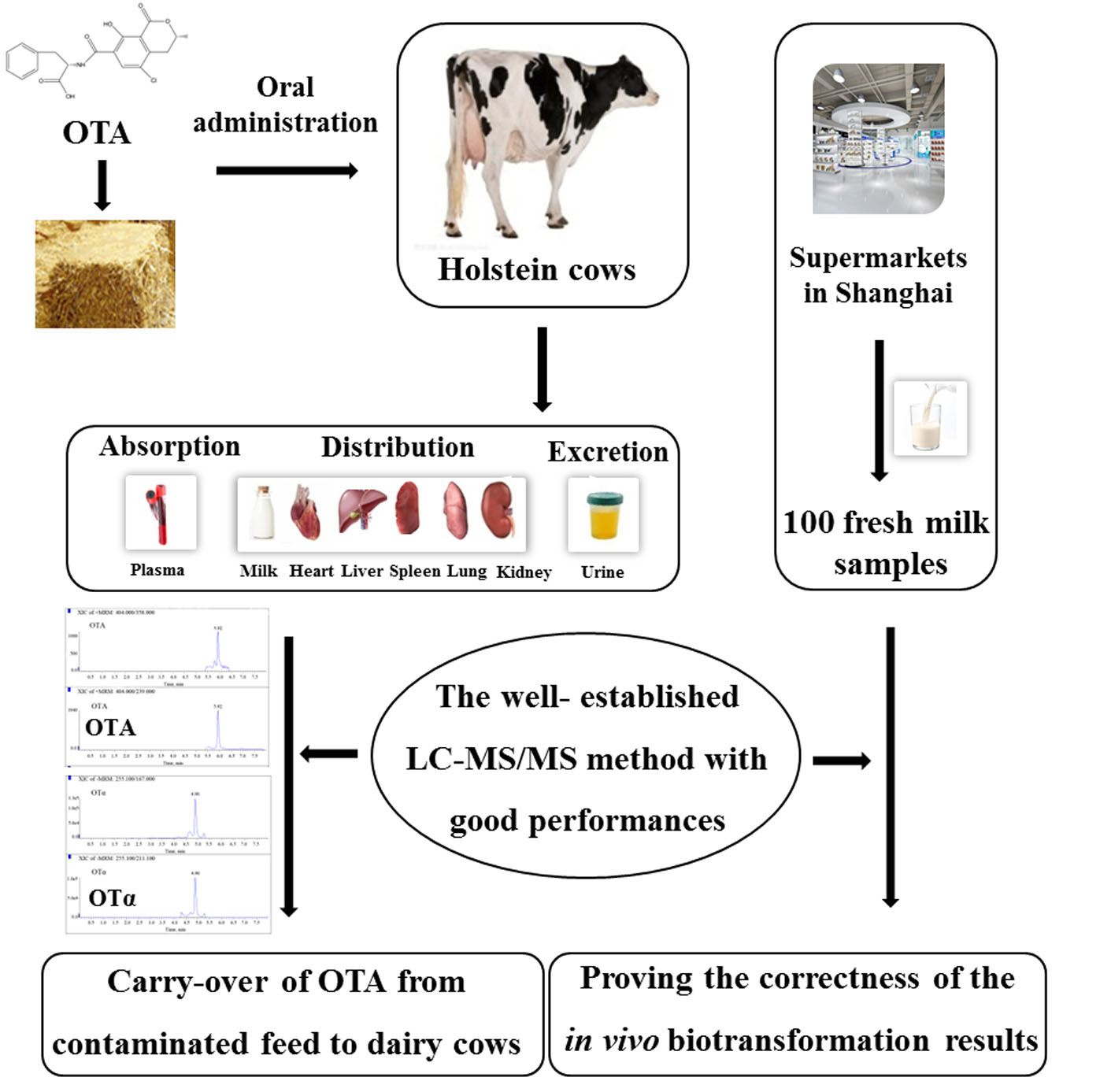
:
1. Introduction
2. Results
2.1. Optimization of Extraction Solvent
2.2. LC-MS/MS Method Validation
2.3. Method Application
3. Discussion
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. Apparatus
4.3. Diets and Animals
4.4. Sample Collection
4.5. Sample Pretreatment
4.6. LC-MS/MS Method Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| b.w. | Body weight |
| CONTAM | Contaminants in the Food Chain |
| ELISA | Enzyme-linked immunosorbent assay |
| EFSA | European Food Safety Authority |
| FLD | Fluorescence detector |
| HPLC | High-performance liquid chromatography |
| HPLC-MS/MS | High-performance liquid chromatography-tandem mass spectrometry |
| HRMS | High-resolution mass spectrometry |
| IARC | International Agency for Research on Cancer |
| JECFA | Joint FAO/WHO Expert Committee on Food Additives |
| LC-MS/MS | Liquid chromatography-tandem mass spectrometry |
| LLOD | Lower limit of detection |
| LLOQ | Lower limit of quantification |
| MRM | Multiple reaction monitoring |
| MS | Mass spectrometry |
| NMR-MS | Nuclear magnetic resonance and mass spectrometry |
| OTA | Ochratoxin A |
| OTα | Ochratoxin α |
| PTWI | Provisional tolerable weekly intake |
| RT | Room temperature |
| SSE | Signal suppression/enhancement |
| TLC | Thin-layer chromatography |
| TMR | Total mixed rations |
| MS/MS | Tandem mass spectrometry |
| TWI | Tolerable Weekly Intake |
References
- Malir, F.; Ostry, V.; Pfohl-Leszkowicz, A.; Malir, J.; Toman, J. Ochratoxin A: 50 years of research. Toxins 2016, 8, 191. [Google Scholar] [CrossRef] [PubMed]
- Gil-Serna, J.; Garcia-Diaz, M.; Vazquez, C.; Gonzalez-Jaen, M.T.; Patino, B. Significance of Aspergillus niger aggregate species as contaminants of food products in Spain regarding their occurrence and their ability to produce mycotoxins. Food Microbiol. 2019, 82, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Van Der Merwe, K.J.; Steyn, P.S.; Fourie, L.; Scott, D.B.; Theron, J.J. Ochratoxin A, a toxic metabolite produced by Aspergillus ochraceus Wilh. Nature 1965, 205, 1112–1113. [Google Scholar] [CrossRef] [PubMed]
- Pleadin, J.; Kudumija, N.; Kovacevic, D.; Scortichini, G.; Milone, S.; Kmetic, I. Comparison of ochratoxin A levels in edible pig tissues and in biological fluids after exposure to a contaminated diet. Mycotoxin Res. 2016, 32, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Escriva, L.; Font, G.; Manyes, L.; Berrada, H. Studies on the presence of mycotoxins in biological samples: An overview. Toxins 2017, 9, 251. [Google Scholar] [CrossRef] [PubMed]
- EFSA. Opinion of the scientific panel on contaminants in the food chain on a request from the commission related to ochratoxin A in food. EFSA J. 2006, 365, 1–56. [Google Scholar] [CrossRef]
- Vettorazzi, A.; Gonzalez-Penas, E.; de Cerain, A.L. Ochratoxin A kinetics: A review of analytical methods and studies in rat model. Food Chem. Toxicol. 2014, 72, 273–288. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Katsunuma, Y.; Nunokawa, M.; Minato, H.; Yonemochi, C. Influence of repeated ochratoxin A ingestion on milk production and its carry-over into the milk, blood and tissues of lactating cows. Anim. Sci. J. 2016, 87, 541–546. [Google Scholar] [CrossRef]
- Han, Z.; Zhao, Z.; Shi, J.; Liao, Y.; Zhao, Z.; Zhang, D.; Wu, Y.; De Saeger, S.; Wu, A. Combinatorial approach of LC–MS/MS and LC–TOF-MS for uncovering in vivo kinetics and biotransformation of ochratoxin A in rat. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 925, 46–53. [Google Scholar] [CrossRef]
- Shreeve, B.J.; Patterson, D.S.P.; Roberts, B.A. The ‘carry-over’ of aflatoxin, ochratoxin and zearalenone from naturally contaminated feed to tissues, urine and milk of dairy cows. Food Cosmet. Toxicol. 1979, 17, 151–152. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, H.; Saeger, S.D.; Boevre, M.D.; Sun, F.; Zhang, S.; Cao, X.; Wang, Z. In vitro and in vivo metabolism of ochratoxin A: A comparative study using ultra-performance liquid chromatography- quadrupole/time-of-flight hybrid mass spectrometry. Anal. Bioanal. Chem. 2015, 407, 3579–3589. [Google Scholar] [CrossRef]
- Vettorazzi, A.; de Trocóniz, I.F.; González-Peñas, E.; Arbillaga, L.; Corcuera, L.A.; Gil, A.G.; de Cerain, A.L. Kidney and liver distribution of ochratoxin A in male and female F344 rats. Food Chem. Toxicol. 2011, 49, 1935–1942. [Google Scholar] [CrossRef]
- Mobashar, M.; Hummel, J.; Blank, R.; Südekum, K.H. Ochratoxin A in ruminants—A review on its degradation by gut microbes and effects on animals. Toxins 2010, 2, 809–839. [Google Scholar] [CrossRef] [PubMed]
- Müller, H.M.; Lerch, C.; Müller, K.; Eggert, W. Kinetic profiles of ochratoxin A and ochratoxin alpha during in vitro incubation in buffered forestomach and abomasal contents from cows. Nat. Toxins 1998, 6, 251–258. [Google Scholar] [CrossRef]
- Mobashar, M. Ruminal Degradation of Ochratoxin A—In Vitro Investigations at Varying Rations and Rumen Microbial Populations; Faculty of Agriculture Rheinische Friedrich-Wilhelms-University: Bonn, Germany, 2011. [Google Scholar]
- Ribelin, W.E.; Fukushima, K.; Still, P.E. The toxicity of ochratoxin to ruminants. Can. J. Comp. Med. 1978, 42, 172. [Google Scholar] [PubMed]
- Sreemannarayana, O.; Frohlich, A.A.; Vitti, T.G.; Marquardt, R.R.; Abramson, D. Studies of the tolerance and disposition of ochratoxin A in young calves. J. Anim. Sci. 1988, 66, 1703–1711. [Google Scholar] [CrossRef] [PubMed]
- Scott, P.M.; Lawrence, J.W.; van Walbeek, W. Detection of mycotoxins by thin-layer chromatography: Application to screening of fungal extracts. Appl. Microbiol. 1970, 20, 839–842. [Google Scholar] [CrossRef]
- Santos, E.A.; Vargas, E.A. Immunoaffinity column clean-up and thin layer chromatography for determination of ochratoxin A in green coffee. Food Addit. Contam. 2002, 19, 447–458. [Google Scholar] [CrossRef]
- Eshelli, M.; Qader, M.M.; Jambi, E.J.; Hursthouse, A.S.; Rateb, M.E. Current status and future opportunities of omics tools in mycotoxin research. Toxins 2018, 10, 433. [Google Scholar] [CrossRef]
- Lauwers, M.; De Baere, S.; Letor, B.; Rychlik, M.; Croubels, S.; Devreese, M. Multi LC-MS/MS and LC- HRMS methods for determination of 24 mycotoxins including major phase I and II biomarker metabolites in biological matrices from pigs and broiler chickens. Toxins 2019, 11, 171. [Google Scholar] [CrossRef]
- FAO. Dairy Animals. Available online: http://www.fao.org/agriculture/dairy-gateway/milkproduction/dairy-animals/en/#.VS6myfB5JOY (accessed on 5 April 2015).
- Pattono, D.; Gallo, P.F.; Civera, T. Detection and quantification of ochratoxin A in milk produced in organic farms. Food Chem. 2011, 127, 374–377. [Google Scholar] [CrossRef]
- Boudra, H.; Barnouin, J.; Dragacci, S.; Morgavi, D.P. Aflatoxin M1 and ochratoxin A in raw bulk milk from French dairy herds. J. Dairy Sci. 2007, 90, 3197–3201. [Google Scholar] [CrossRef]
- Galtier, P. Pharmacokinetics of ochratoxin A in Animals. IARC Sci. Publ. 1991, 115, 187–200. [Google Scholar]
- Boudra, H.; Saivin, S.; Buffiere, C.; Morgavi, D.P. Short communication: Toxicokinetics of ochratoxin A in dairy ewes and carryover to milk following a single or long-term ingestion of contaminated feed. J. Dairy Sci. 2013, 96, 6690–6696. [Google Scholar] [CrossRef] [Green Version]
- Müller, H.M.; Müller, K.; Steingass, H. Effect of feeding regime on the metabolism of ochratoxin a during the in vitro incubation in buffered rumen fluid from cows. Arch Tierernahr. 2001, 54, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Mobashar, M.; Blank, R.; Hummel, J.; Westphal, A.; Tholen, E.; Südekum, K.H. Ruminal ochratoxin A degradation-contribution of the different microbial populations and influence of diet. Anim. Feed Sci. Tech. 2012, 171, 85–97. [Google Scholar] [CrossRef]
- Pattono, D.; Grosso, A.; Stocco, P.P.; Pazzi, M.; Zeppa, G. Survey of the presence of patulin and ochratoxin A in traditional semi-hard cheeses. Food Control 2013, 33, 54–57. [Google Scholar] [CrossRef]
Sample Availability: Not available. |


{kind=link}
{kind=link}
| Matrix | Mycotoxin | Linear Range (ng mL−1) | Slope | Intercept | R2 | LLOD (ng mL−1) | LLOQ (ng mL−1) | SSE (%) |
|---|---|---|---|---|---|---|---|---|
| Solvent | OTA | 0.1–200 | 12,601 | 275 | 0.999 | / | / | / |
| OTα | 0.1–200 | 37,500 | 846 | 0.999 | / | / | / | |
| Milk | OTA | 0.1–200 | 20,432 | 542 | 0.994 | 0.03 | 0.1 | 162 |
| OTα | 0.1–200 | 21,438 | 1215 | 0.999 | 0.03 | 0.1 | 57 | |
| Urine | OTA | 0.2–200 | 14,564 | 180 | 0.990 | 0.1 | 0.2 | 116 |
| OTα | 0.2–200 | 13,428 | 197 | 0.990 | 0.1 | 0.2 | 36 | |
| Plasma | OTA | 0.1–200 | 26,422 | −2142 | 0.995 | 0.03 | 0.1 | 130 |
| OTα | 0.1–200 | 27,476 | 2219 | 0.997 | 0.03 | 0.1 | 73 | |
| Heart | OTA | 0.1–200 | 6378 | 810 | 0.997 | 0.03 | 0.1 | 51 |
| OTα | 0.1–200 | 30,231 | 2037 | 0.994 | 0.03 | 0.1 | 81 | |
| Liver | OTA | 0.1–200 | 5598 | 1109 | 0.999 | 0.03 | 0.1 | 44 |
| OTα | 0.1–200 | 24,216 | −501 | 0.997 | 0.03 | 0.1 | 65 | |
| Spleen | OTA | 0.1–200 | 4990 | 636 | 0.998 | 0.03 | 0.1 | 40 |
| OTα | 0.1–200 | 26,424 | 1424 | 0.993 | 0.03 | 0.1 | 70 | |
| Lung | OTA | 0.1–200 | 4934 | 617 | 0.999 | 0.03 | 0.1 | 39 |
| OTα | 0.1–200 | 22,983 | 1086 | 0.998 | 0.03 | 0.1 | 61 | |
| Kidney | OTA | 0.1–200 | 5048 | 851 | 0.998 | 0.03 | 0.1 | 40 |
| OTα | 0.1–200 | 26,374 | 700 | 0.995 | 0.03 | 0.1 | 70 |
| Matrix | Mycotoxin | Spiked Level (ng mL−1) | Recovery (Mean ± SD, %) | Intra-Day Precision (RSD, %) | Inter-Day Precision (RSD, %) |
|---|---|---|---|---|---|
| Milk | OTA | LLOQ | 78.6 ± 4.0 | 5.0 | 10.7 |
| 1 | 95.4 ± 10.7 | 11.2 | 12.0 | ||
| 50 | 86.3 ± 5.8 | 6.7 | 11.0 | ||
| 200 | 80.1 ± 8.1 | 10.1 | 11.3 | ||
| OTα | LLOQ | 106.7 ± 5.3 | 4.9 | 12.2 | |
| 1 | 88.9 ± 9.4 | 10.6 | 11.9 | ||
| 50 | 94.6 ± 8.0 | 8.5 | 12.1 | ||
| 200 | 100.3 ± 3.5 | 3.5 | 8.6 | ||
| Urine | OTA | LLOQ | 75.3 ± 4.1 | 5.5 | 12.6 |
| 1 | 82.4 ± 5.2 | 6.3 | 11.3 | ||
| 50 | 87.4 ± 6.1 | 6.9 | 10.8 | ||
| 200 | 88.7 ± 6.0 | 6.8 | 9.9 | ||
| OTα | LLOQ | 99.3 ± 6.4 | 6.5 | 10.5 | |
| 1 | 83.8 ± 8.1 | 9.6 | 12.2 | ||
| 50 | 79.3 ± 5.8 | 7.3 | 9.0 | ||
| 200 | 84.3 ± 6.8 | 8.1 | 11.7 | ||
| Plasma | OTA | LLOQ | 87.9 ± 9.7 | 11.0 | 13.4 |
| 1 | 84.9 ± 7.3 | 8.6 | 11.3 | ||
| 50 | 83.5 ± 8.8 | 10.6 | 10.6 | ||
| 200 | 75.6 ± 4.5 | 5.9 | 10.1 | ||
| OTα | LLOQ | 114.1 ± 2.2 | 1.9 | 10.3 | |
| 1 | 87.9 ± 8.6 | 9.8 | 12.9 | ||
| 50 | 97.3 ± 4.4 | 4.5 | 6.1 | ||
| 200 | 92.6 ± 11.8 | 12.8 | 13.6 | ||
| Heart | OTA | LOQ | 112.1 ± 4.9 | 4.4 | 8.6 |
| 1 | 94.0 ± 7.5 | 8.0 | 11.9 | ||
| 50 | 91.1 ± 9.4 | 10.4 | 11.8 | ||
| 200 | 99.0 ± 3.6 | 3.7 | 8.6 | ||
| OTα | LLOQ | 101.1 ± 5.9 | 5.8 | 12.4 | |
| 1 | 87.0 ± 9.7 | 11.2 | 12.0 | ||
| 50 | 96.8 ± 9.0 | 9.3 | 11.7 | ||
| 200 | 89.2 ± 9.1 | 10.2 | 10.3 | ||
| Liver | OTA | LLOQ | 101.1 ± 6.3 | 6.3 | 10.2 |
| 1 | 91.0 ± 6.5 | 7.1 | 10.3 | ||
| 50 | 102.1 ± 6.8 | 6.7 | 11.9 | ||
| 200 | 91.9 ± 9.7 | 10.6 | 12.3 | ||
| OTα | LLOQ | 81.6 ± 6.9 | 8.4 | 11.6 | |
| 1 | 94.7 ± 5.2 | 5.5 | 9.7 | ||
| 50 | 102.8 ± 5.0 | 4.9 | 9.3 | ||
| 200 | 95.4 ± 4.3 | 4.5 | 10.8 | ||
| Spleen | OTA | LLOQ | 109.8 ± 8.0 | 7.3 | 7.9 |
| 1 | 97.8 ± 6.3 | 6.5 | 12.5 | ||
| 50 | 91.1 ± 2.1 | 2.3 | 11.4 | ||
| 200 | 96.7 ± 5.6 | 5.8 | 10.4 | ||
| OTα | LLOQ | 105.6 ± 4.3 | 4.1 | 12.1 | |
| 1 | 99.4 ± 8.9 | 8.9 | 9.7 | ||
| 50 | 93.0 ± 6.5 | 7.0 | 12.0 | ||
| 200 | 90.1 ± 6.4 | 7.1 | 10.9 | ||
| Lung | OTA | LLOQ | 99.0 ± 4.9 | 5.0 | 8.0 |
| 1 | 89.6 ± 9.1 | 10.2 | 11.6 | ||
| 50 | 89.0 ± 8.0 | 8.9 | 9.8 | ||
| 200 | 85.3 ± 5.0 | 5.8 | 10.1 | ||
| OTα | LLOQ | 107.5 ± 6.4 | 5.9 | 10.5 | |
| 1 | 97.7 ± 3.8 | 3.9 | 9.4 | ||
| 50 | 94.0 ± 5.7 | 6.0 | 11.4 | ||
| 200 | 98.0 ± 5.8 | 5.9 | 11.6 | ||
| Kidney | OTA | LLOQ | 105.2 ± 8.0 | 7.6 | 12.6 |
| 1 | 91.0 ± 5.4 | 6.0 | 11.2 | ||
| 50 | 99.5 ± 2.5 | 2.6 | 10.9 | ||
| 200 | 88.1 ± 9.1 | 10.4 | 12.8 | ||
| OTα | LLOQ | 108.9 ± 6.3 | 5.8 | 10.8 | |
| 1 | 90.9 ± 10.7 | 11.8 | 11.6 | ||
| 50 | 94.1 ± 7.8 | 8.3 | 12.9 | ||
| 200 | 87.4 ± 8.7 | 9.9 | 10.2 |
| Matrix | Mycotoxin | Spiked Level (ng mL−1) | Recovery (Mean ± SD %) | ||
|---|---|---|---|---|---|
| Short-Term Stability a | Freeze-Thaw Stability b | Long-Term Stability c | |||
| Urine | OTA | 1 | 98.3 ± 8.1 | 97.6 ± 6.1 | 98.7 ± 6.7 |
| 50 | 101.4 ± 2.4 | 102.2 ± 4.9 | 99.8 ± 3.6 | ||
| OTα | 1 | 89.6 ± 2.9 | 88.3 ± 7.9 | 87.9 ± 8.1 | |
| 50 | 98.5 ± 7.0 | 93.6 ± 4.6 | 92.3 ± 4.8 | ||
| Plasma | OTA | 1 | 86.0 ± 7.8 | 113.6 ± 2.8 | 93.4 ± 7.4 |
| 50 | 90.7 ± 11.8 | 93.2 ± 7.3 | 86.7 ± 6.7 | ||
| OTα | 1 | 96.2 ± 11.7 | 94.7 ± 8.5 | 96.9 ± 7.8 | |
| 50 | 88.6 ± 5.5 | 96.7 ± 9.3 | 94.4 ± 4.6 | ||
| Heart | OTA | 1 | 90.5 ± 8.6 | 95.3 ± 8.6 | 91.1 ± 5.1 |
| 50 | 85.8 ± 7.1 | 91.3 ± 9.1 | 99.0 ± 11.9 | ||
| OTα | 1 | 99.0 ± 8.4 | 86.3 ± 10.4 | 95.5 ± 12.4 | |
| 50 | 91.3 ± 11.6 | 90.8 ± 7.2 | 97.1 ± 8.6 | ||
| Liver | OTA | 1 | 91.0 ± 9.4 | 98.0 ± 10.1 | 93.5 ± 9.5 |
| 50 | 90.5 ± 8.2 | 94.2 ± 10.0 | 92.1 ± 10.3 | ||
| OTα | 1 | 92.4 ± 7.2 | 92.6 ± 10.4 | 98.3 ± 9.4 | |
| 50 | 95.3 ± 8.5 | 93.3 ± 6.8 | 98.9 ± 9.6 | ||
| Spleen | OTA | 1 | 87.7 ± 12.3 | 84.3 ± 6.1 | 90.0 ± 9.9 |
| 50 | 96.8 ± 10.1 | 88.8 ± 5.1 | 97.9 ± 5.5 | ||
| OTα | 1 | 92.0 ± 12.6 | 96.2 ± 12.1 | 83.0 ± 5.1 | |
| 50 | 94.4 ± 6.1 | 98.5 ± 9.8 | 92.5 ± 9.4 | ||
| Lung | OTA | 1 | 86.1 ± 8.1 | 92.9 ± 9.7 | 93.3 ± 9.8 |
| 50 | 90.9 ± 7.7 | 97.0 ± 11.4 | 85.1 ± 3.9 | ||
| OTα | 1 | 89.6 ± 8.2 | 94.4 ± 10.9 | 98.1 ± 8.3 | |
| 50 | 93.7 ± 9.8 | 96.9 ± 10.2 | 85.3 ± 10.8 | ||
| Kidney | OTA | 1 | 87.8 ± 9.4 | 87.3 ± 9.1 | 93.8 ± 6.0 |
| 50 | 100.4 ± 7.4 | 86.6 ± 8.7 | 90.6 ± 10.0 | ||
| OTαα | 1 | 89.3 ± 9.7 | 97.2 ± 8.1 | 92.7 ± 9.7 | |
| 50 | 98.2 ± 8.3 | 91.8 ± 8.9 | 99.0 ± 6.0 | ||
| Time (min) | OTA | OTα |
|---|---|---|
| 120 | - | - |
| 360 | 1.6 ± 0.12 | 203.8 ± 16.3 |
| 720 | 1.8 ± 0.09 | 324.6 ± 23.2 |
| 1440 | 1.0 ± 0.06 | 232.6 ± 13.9 |
| 2160 | 0.6 ± 0.07 | 141.2 ± 11.3 |
| 2880 | - | 84.2 ± 7.6 |
| 4320 | - | - |
| Animals | Dosage | Mycotoxin/Administration | Matrices | Analytical Method | LLOD (ng mL−1) | LLOQ (ng mL−1) | Results | Reference | ||
|---|---|---|---|---|---|---|---|---|---|---|
| OTA | OTα | OTA | OTα | |||||||
| Jersey milking cows | 317–1125 μg/kg diets | Naturally contaminated barley containing OTA/Feeding | Milk, serum, urine, and tissues a | TLC | 5 | 5 | / | / | OTA(5 μg/kg) was detected in kidney | [10] |
| Holstein cows | 0.2–13.3 mg/kg b.w. | Pure OTA/By stomach | Milk and urine | TLC | / | / | / | / | OTA and OTα were detected in milk and urine d | [16] |
| Young Holstein-Friesian male calves | 0.25–2 mg/kg b.w. | Pure OTA/Feeding | Serum, urine, and feces | HPLC | 50 | 50 | / | / | OTA and OTα were detected in serum, urine and feces e | [17] |
| Holstein cows | 5, 50 or 100 μg/kg diets | Pure OTA/Feeding | Plasma, milk, and tissues b | HPLC | / | / | 0.1 | / | OTA (0.1 μg/kg) was detected in the plasma | [8] |
| Holstein cows | 30 μg/kg b.w. | Pure OTA/ Feeding | Plasma, milk, urine, and tissues c | HPLC-MS/MS | 0.03–0.1 | 0.03–0.1 | 0.1–0.2 | 0.1–0.2 | OTA and OTα were detected in urine | This study |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Fan, Z.; Nie, D.; Zhao, Z.; Han, Z. Analysis of the Carry-Over of Ochratoxin A from Feed to Milk, Blood, Urine, and Different Tissues of Dairy Cows Based on the Establishment of a Reliable LC-MS/MS Method. Molecules 2019, 24, 2823. https://doi.org/10.3390/molecules24152823
Zhang Z, Fan Z, Nie D, Zhao Z, Han Z. Analysis of the Carry-Over of Ochratoxin A from Feed to Milk, Blood, Urine, and Different Tissues of Dairy Cows Based on the Establishment of a Reliable LC-MS/MS Method. Molecules. 2019; 24(15):2823. https://doi.org/10.3390/molecules24152823
Chicago/Turabian StyleZhang, Zhiqi, Zhichen Fan, Dongxia Nie, Zhihui Zhao, and Zheng Han. 2019. "Analysis of the Carry-Over of Ochratoxin A from Feed to Milk, Blood, Urine, and Different Tissues of Dairy Cows Based on the Establishment of a Reliable LC-MS/MS Method" Molecules 24, no. 15: 2823. https://doi.org/10.3390/molecules24152823
APA StyleZhang, Z., Fan, Z., Nie, D., Zhao, Z., & Han, Z. (2019). Analysis of the Carry-Over of Ochratoxin A from Feed to Milk, Blood, Urine, and Different Tissues of Dairy Cows Based on the Establishment of a Reliable LC-MS/MS Method. Molecules, 24(15), 2823. https://doi.org/10.3390/molecules24152823