Vancomycin-Iridium (III) Interaction: An Unexplored Route for Enantioselective Imine Reduction

 , , ,
, , ,  and
and
Abstract
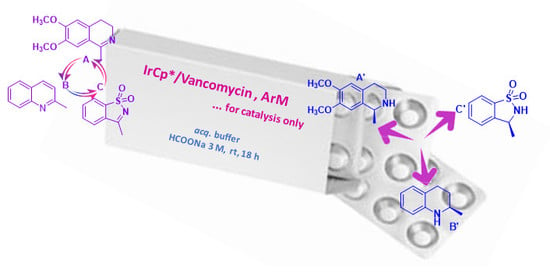
:
1. Experimental Section
1.1. General Procedure for Synthesis of [Ir(Cp*)(Van)Cl] Complex
1.2. General Procedure for Asymmetric Transfer Hydrogenation.
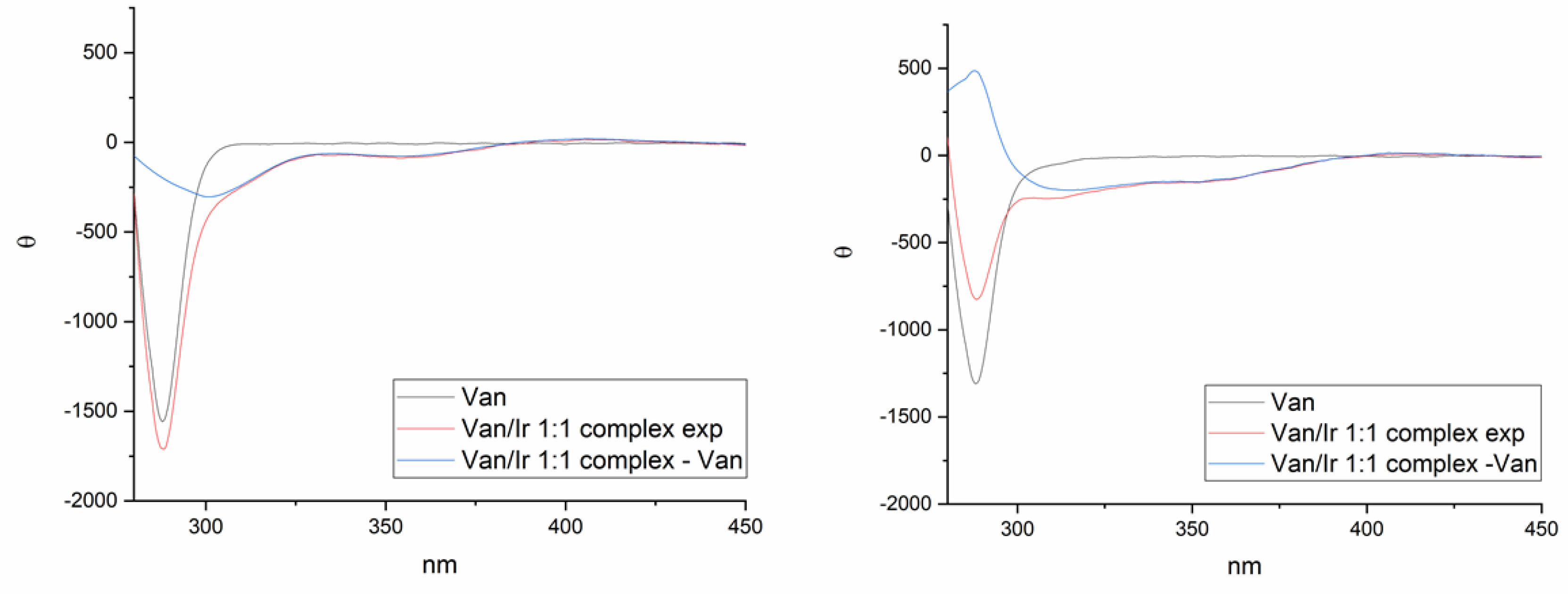
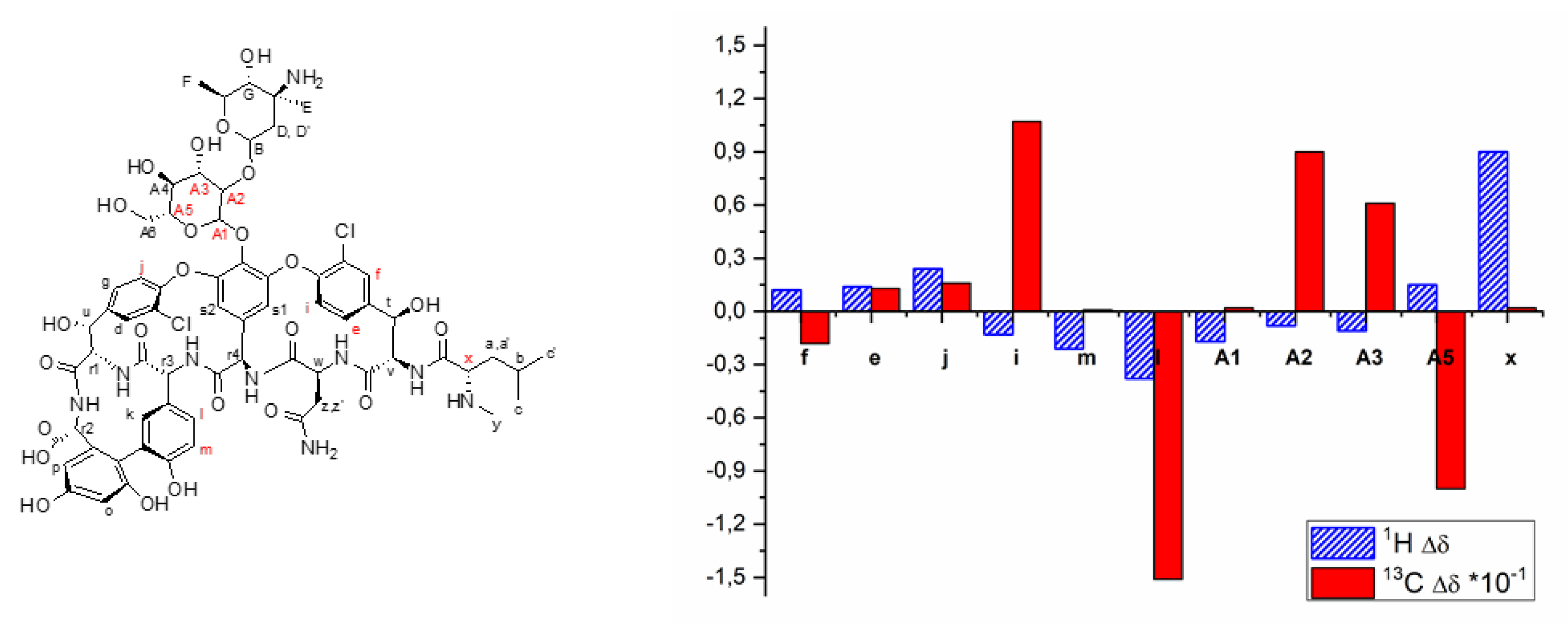
1.3. Circular Dichroism
1.4. Kinetic Experiments
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Xing, B.; Jiang, T.; Wu, X.; Liew, R.; Zhou, J.; Zhang, D.; Yeow, E.K.L. Molecular Interactions between Glycopeptide Vancomycin and Bacterial Cell Wall Peptide Analogues. Chem. Eur. J. 2011, 17, 14170–14177. [Google Scholar] [CrossRef] [PubMed]
- Nitanai, Y.; Kikuchi, T.; Kakoi, K.; Hanamaki, S.; Fujisawa, I.; Aoki, K. Crystal Structures of the Complexes between Vancomycin and Cell-Wall Precursor Analogs. J. Mol. Biol. 2009, 385, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
- Świątek, M.; Valensin, D.; Migliorini, C.; Gaggelli, E.; Valensin, G.; Jeżowska-Bojczuk, M. Unusual binding ability of vancomycin towards Cu2+ ions. Dalton Trans. 2005, 23, 3808–3813. [Google Scholar] [CrossRef]
- Nair, U.B.; Chang, S.S.C.; Armstrong, D.W.; Rawjee, Y.Y.; Eggleston, D.S.; McArdle, J.V. Elucidation of vancomycin’s enantioselective binding site using its copper complex. Chirality 1996, 8, 590–595. [Google Scholar] [CrossRef]
- Zarkan, A.; Macklyne, H.-R.; Truman, A.W.; Hesketh, A.R.; Hong, H.-J. The frontline antibiotic vancomycin induces a zinc starvation response in bacteria by binding to Zn(II). Sci. Rep. 2016, 6, 19602. [Google Scholar] [CrossRef] [PubMed]
- Fusè, M.; Rimoldi, I.; Cesarotti, E.; Rampino, S.; Barone, V. On the relation between carbonyl stretching frequencies and the donor power of chelating diphosphines in nickel dicarbonyl complexes. Phys. Chem. Chem. Phys. 2017, 19, 9028–9038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facchetti, G.; Cesarotti, E.; Pellizzoni, M.; Zerla, D.; Rimoldi, I. “In situ” Activation of Racemic RuII Complexes: Separation of trans and cis Species and Their Application in Asymmetric Reduction. Eur. J. Inorg. Chem. 2012, 2012, 4365–4370. [Google Scholar] [CrossRef]
- Fusè, M.; Rimoldi, I.; Facchetti, G.; Rampino, S.; Barone, V. Exploiting coordination geometry to selectively predict the σ-donor and π-acceptor abilities of ligands: A back-and-forth journey between electronic properties and spectroscopy. Chem. Commun. 2018, 54, 2397–2400. [Google Scholar] [CrossRef]
- Schäfer, M.; Schneider, T.R.; Sheldrick, G.M. Crystal structure of vancomycin. Structure 1996, 4, 1509–1515. [Google Scholar] [CrossRef]
- Dieguez, M.; Backvall, J.E.; Pamies, O. Artificial Metalloenzymes and MetalloDNAzymes in Catalysis: From Design to Applications, 1st ed.; WILEY-VCH: Weinheim, Germany, 2018; p. 432. [Google Scholar]
- Jeschek, M.; Panke, S.; Ward, T.R. Artificial Metalloenzymes on the Verge of New-to-Nature Metabolism. Trends Biotechnol. 2018, 36, 60–72. [Google Scholar] [CrossRef] [Green Version]
- Schwizer, F.; Okamoto, Y.; Heinisch, T.; Gu, Y.; Pellizzoni, M.M.; Lebrun, V.; Reuter, R.; Köhler, V.; Lewis, J.C.; Ward, T.R. Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 2018, 118, 142–231. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, S.; Facchetti, G.; Contini, A.; Gelmi, M.L.; Erba, E.; Gandolfi, R.; Rimoldi, I. Ctr-1 Mets7 motif inspiring new peptide ligands for Cu(i)-catalyzed asymmetric Henry reactions under green conditions. RSC Adv. 2016, 6, 71529–71533. [Google Scholar] [CrossRef] [Green Version]
- Filice, M.; Romero, O.; Gutiérrez-Fernández, J.; de las Rivas, B.; Hermoso, J.A.; Palomo, J.M. Synthesis of a heterogeneous artificial metallolipase with chimeric catalytic activity. Chem. Commun. 2015, 51, 9324–9327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilie, A.; Reetz, M.T. Directed Evolution of Artificial Metalloenzymes. Isr. J. Chem. 2015, 55, 51–60. [Google Scholar] [CrossRef]
- Sreenilayam, G.; Moore, E.J.; Steck, V.; Fasan, R. Metal Substitution Modulates the Reactivity and Extends the Reaction Scope of Myoglobin Carbene Transfer Catalysts. Adv. Synth. Catal. 2017, 359, 2076–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, J.C. Artificial Metalloenzymes and Metallopeptide Catalysts for Organic Synthesis. ACS Catal. 2013, 3, 2954–2975. [Google Scholar] [CrossRef]
- Zhu, R.; Xu, Z.; Ding, W.; Liu, S.; Shi, X.; Lu, X. Efficient and Practical Syntheses of Enantiomerically Pure (S)-(−)-Norcryptostyline I, (S)-(−)-Norcryptostyline II, (R)-(+)-Salsolidine and (S)-(−)-Norlaudanosine via a Resolution-Racemization Method. Chin. J. Chem. 2014, 32, 1039–1048. [Google Scholar] [CrossRef]
- Bucci, R.; Bonetti, A.; Clerici, F.; Contini, A.; Nava, D.; Pellegrino, S.; Tessaro, D.; Gelmi, M.L. Tandem Tetrahydroisoquinoline-4-carboxylic Acid/β-Alanine as a New Construct Able To Induce a Flexible Turn. Chem. Eur. J. 2017, 23, 10822–10831. [Google Scholar] [CrossRef] [PubMed]
- Farina, V.; Reeves, J.T.; Senanayake, C.H.; Song, J.J. Asymmetric Synthesis of Active Pharmaceutical Ingredients. Chem. Rev. 2006, 106, 2734–2793. [Google Scholar] [CrossRef]
- Debnath, S.; Mondal, S. Sultams: Recent Syntheses and Applications. Eur. J. Org. Chem. 2018, 2018, 933–956. [Google Scholar] [CrossRef]
- Vázquez-Villa, H.; Reber, S.; Ariger, M.A.; Carreira, E.M. Iridium Diamine Catalyst for the Asymmetric Transfer Hydrogenation of Ketones. Angew. Chem. Int. Ed. 2011, 50, 8979–8981. [Google Scholar] [CrossRef] [PubMed]
- Rimoldi, I.; Facchetti, G.; Cesarotti, E.; Pellizzoni, M.; Fuse, M.; Zerla, D. Enantioselective transfer hydrogenation of aryl ketones: synthesis and 2D-NMR characterization of new 8-amino-5,6,7,8-tetrahydroquinoline Ru(II)-complexes. Curr. Org. Chem. 2012, 16, 2982–2988. [Google Scholar] [CrossRef]
- Lin, Z.; Li, J.; Huang, Q.; Huang, Q.; Wang, Q.; Tang, L.; Gong, D.; Yang, J.; Zhu, J.; Deng, J. Chiral Surfactant-Type Catalyst: Enantioselective Reduction of Long-Chain Aliphatic Ketoesters in Water. J. Org. Chem. 2015, 80, 4419–4429. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Aboo, A.H.; Robertson, C.M.; Liu, R.; Li, Z.; Luzyanin, K.; Berry, N.G.; Chen, W.; Xiao, J. N,O- vs. N,C-Chelation in Half-Sandwich Iridium Complexes: A Dramatic Effect on Enantioselectivity in Asymmetric Transfer Hydrogenation of Ketones. ACS Catal. 2018, 8, 8020–8026. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, T.; Liu, X.; Shen, L. Enantioselective Synthesis of (S)-γ-Amino Alcohols by Ru/Rh/Ir Catalyzed Asymmetric Transfer Hydrogenation (ATH) with Tunable Chiral Tetraaza Ligands in Water. Catal. Lett. 2019, 1–9. [Google Scholar] [CrossRef]
- Foubelo, F.; Nájera, C.; Yus, M. Catalytic asymmetric transfer hydrogenation of ketones: recent advances. Tetrahedron Asymmetry 2015, 26, 769–790. [Google Scholar] [CrossRef] [Green Version]
- Václavík, J.K.; Kacer, P.; Kuzma, M.; Červený, L. Opportunities Offered by Chiral η6-Arene/N-Arylsulfonyl-diamine-RuII Catalysts in the Asymmetric Transfer Hydrogenation of Ketones and Imines. Molecules 2011, 16, 5460–5495. [Google Scholar] [CrossRef]
- Facchetti, G.; Bucci, R.; Fusè, M.; Rimoldi, I. Asymmetric Hydrogenation vs. Transfer Hydrogenation in the Reduction of Cyclic Imines. ChemistrySelect 2018, 3, 8797–8800. [Google Scholar] [CrossRef]
- Vilhanova, B.; Vaclavik, J.; Sot, P.; Pechacek, J.; Zapal, J.; Pazout, R.; Maixner, J.; Kuzma, M.; Kacer, P. Enantioselective hydrogenation of cyclic imines catalysed by Noyori-Ikariya half-sandwich complexes and their analogues. Chem. Commun. 2016, 52, 362–365. [Google Scholar] [CrossRef]
- Itsuno, S.; Hashimoto, Y.; Haraguchi, N. Synthesis of chiral iridium complexes immobilized on amphiphilic polymers and their application to asymmetric catalysis. J. Polym. Sci. Part A: Polym. Chem. 2014, 52, 3037–3044. [Google Scholar] [CrossRef]
- Ikariya, T.; Murata, K.; Noyori, R. Bifunctional transition metal-based molecular catalysts for asymmetric syntheses. Org. Biomol. Chem. 2006, 4, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Facchetti, G.; Rimoldi, I. 8-Amino-5,6,7,8-tetrahydroquinoline in iridium(iii) biotinylated Cp* complex as artificial imine reductase. New J. Chem. 2018, 42, 18773–18776. [Google Scholar] [CrossRef]
- Pellizzoni, M.; Facchetti, G.; Gandolfi, R.; Fusè, M.; Contini, A.; Rimoldi, I. Evaluation of Chemical Diversity of Biotinylated Chiral 1,3-Diamines as a Catalytic Moiety in Artificial Imine Reductase. ChemCatChem 2016, 8, 1665–1670. [Google Scholar] [CrossRef] [Green Version]
- Valencia, M.; Müller-Bunz, H.; Gossage, R.A.; Albrecht, M. Enhanced product selectivity promoted by remote metal coordination in acceptor-free alcohol dehydrogenation catalysis. Chem. Commun. 2016, 52, 3344–3347. [Google Scholar] [CrossRef] [Green Version]
- Ricciardi, L.; La Deda, M.; Ionescu, A.; Godbert, N.; Aiello, I.; Ghedini, M.; Fusè, M.; Rimoldi, I.; Cesarotti, E. Luminescent chiral ionic Ir(III) complexes: Synthesis and photophysical properties. J. Lumin. 2016, 170, 812–819. [Google Scholar] [CrossRef]
- McPhail, D.; Cooper, A. Thermodynamics and kinetics of dissociation of ligand-induced dimers of vancomycin antibiotics. J. Chem. Soc. Faraday Trans. 1997, 93, 2283–2289. [Google Scholar] [CrossRef]
- Cicogna, F.; Colonna, M.; Houben, J.L.; Ingrosso, G.; Marchetti, F. Synthesis of 9-anthrylmethyl-functionalised cyclopentadienyl derivatives of rhodium(I) and iridium(I) and study of their luminescence properties. J. Organomet. Chem. 2000, 593–594, 251–266. [Google Scholar] [CrossRef]
- Bertini, F.; Calucci, L.; Cicogna, F.; Gaddi, B.; Ingrosso, G.; Marcaccio, M.; Marchetti, F.; Paolucci, D.; Paolucci, F.; Pinzino, C. Electronic properties of new homobimetallic anthracene-bridged η5-cyclopentadienyl derivatives of iridium(I) and of the corresponding cation radicals [L2Ir{C5H4CH2(9,10-anthrylene)CH2C5H4}IrL2]+. J. Organomet. Chem. 2006, 691, 2987–3002. [Google Scholar] [CrossRef]
- Nejman, P.S.; Morton-Fernandez, B.; Moulding, D.J.; Athukorala Arachchige, K.S.; Cordes, D.B.; Slawin, A.M.Z.; Kilian, P.; Woollins, J.D. Structural diversity of bimetallic rhodium and iridium half sandwich dithiolato complexes. Dalton Trans. 2015, 44, 16758–16766. [Google Scholar] [CrossRef] [Green Version]
- Lora, R.C.; Silveira, L.; Zamuner, S.R.; Pacheco, M.T.T. Dispersive Raman spectroscopy for the in vitro identification and quantification of injected vancomycin intra-vitreous. Spectroscopy 2011, 25, 103–112. [Google Scholar] [CrossRef]
- Ohkuma, T.; Noyori, R. Hydrogenation of imino groups. In Comprehensive Asymmetric Catalysis, Supplement (2004); Jacobsen, E.N., Pfaltz, A., Yamamoto, H., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2004; pp. 43–53. [Google Scholar]
- Cheng, M.; Ziora, Z.M.; Hansford, K.A.; Blaskovich, M.A.; Butler, M.S.; Cooper, M.A. Anti-cooperative ligand binding and dimerisation in the glycopeptide antibiotic dalbavancin. Org. Biomol. Chem. 2014, 12, 2568–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, J.P.; Gerhard, U.; Beauregard, D.A.; Williams, D.H.; Westwell, M.S.; Searle, M.S. Glycopeptide Antibiotic Activity and the Possible Role of Dimerization: A Model for Biological Signaling. J. Am. Chem. Soc. 1994, 116, 4581–4590. [Google Scholar] [CrossRef]
- Rekharsky, M.; Hesek, D.; Lee, M.; Meroueh, S.O.; Inoue, Y.; Mobashery, S. Thermodynamics of Interactions of Vancomycin and Synthetic Surrogates of Bacterial Cell Wall. J. Am. Chem. Soc. 2006, 128, 7736–7737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, C.M.H.; Pinto, I.S.S.; Soares, E.V.; Soares, H.M.V.M. (Un)suitability of the use of pH buffers in biological, biochemical and environmental studies and their interaction with metal ions – a review. RSC Adv. 2015, 5, 30989–31003. [Google Scholar] [CrossRef]
- Aberg, J.B.; Samec, J.S.M.; Backvall, J.-E. Mechanistic investigation on the hydrogenation of imines by [p-(Me2CH)C6H4Me]RuH(NH2CHPhCHPhNSO2C6H4-p-CH3). Experimental support for an ionic pathway. Chem. Commun. 2006, 26, 2771–2773. [Google Scholar] [CrossRef]
- Magee, M.P.; Norton, J.R. Stoichiometric, Catalytic, and Enantioface-Selective Hydrogenation of CN Bonds by an Ionic Mechanism. J. Am. Chem. Soc. 2001, 123, 1778–1779. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Iimura, M.; Magee, M.P.; Norton, J.R.; Zhu, G. Ruthenium-Catalyzed Ionic Hydrogenation of Iminium Cations. Scope and Mechanism. J. Am. Chem. Soc. 2005, 127, 7805–7814. [Google Scholar] [CrossRef]
- Hintermair, U.; Campos, J.; Brewster, T.P.; Pratt, L.M.; Schley, N.D.; Crabtree, R.H. Hydrogen-Transfer Catalysis with Cp*IrIII Complexes: The Influence of the Ancillary Ligands. ACS Catal. 2014, 4, 99–108. [Google Scholar] [CrossRef]
- Dydio, P.; Key, H.M.; Nazarenko, A.; Rha, J.Y.-E.; Seyedkazemi, V.; Clark, D.S.; Hartwig, J.F. An artificial metalloenzyme with the kinetics of native enzymes. Science 2016, 354, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Schwizer, F.; Köhler, V.; Dürrenberger, M.; Knörr, L.; Ward, T.R. Genetic Optimization of the Catalytic Efficiency of Artificial Imine Reductases Based on Biotin–Streptavidin Technology. ACS Catal. 2013, 3, 1752–1755. [Google Scholar] [CrossRef]
- Keller, S.G.; Ringenberg, M.R.; Häussinger, D.; Ward, T.R. Evaluation of the Formate Dehydrogenase Activity of Three-Legged Pianostool Complexes in Dilute Aqueous Solution. Eur. J. Inorg. Chem. 2014, 2014, 5860–5864. [Google Scholar] [CrossRef]
- Dürrenberger, M.; Heinisch, T.; Wilson, Y.M.; Rossel, T.; Nogueira, E.; Knörr, L.; Mutschler, A.; Kersten, K.; Zimbron, M.J.; Pierron, J.; et al. Artificial Transfer Hydrogenases for the Enantioselective Reduction of Cyclic Imines. Angew. Chem. Int. Ed. 2011, 50, 3026–3029. [Google Scholar] [CrossRef] [PubMed]
- Robles, V.M.; Dürrenberger, M.; Heinisch, T.; Lledós, A.; Schirmer, T.; Ward, T.R.; Maréchal, J.-D. Structural, Kinetic, and Docking Studies of Artificial Imine Reductases Based on Biotin–Streptavidin Technology: An Induced Lock-and-Key Hypothesis. J. Am. Chem. Soc. 2014, 136, 15676–15683. [Google Scholar] [CrossRef] [PubMed]
- Zerla, D.; Facchetti, G.; Fuse, M.; Pellizzoni, M.; Castellano, C.; Cesarotti, E.; Gandolfi, R.; Rimoldi, I. 8-Amino-5,6,7,8-tetrahydroquinolines as ligands in iridium(III) catalysts for the reduction of aryl ketones by asymmetric transfer hydrogenation (ATH). Tetrahedron Asymmetry 2014, 25, 1031–1037. [Google Scholar] [CrossRef]
- Johnson, K.A.; Goody, R.S. The Original Michaelis Constant: Translation of the 1913 Michaelis–Menten Paper. Biochemistry 2011, 50, 8264–8269. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 4,5 and 6 are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Buffer | Sub 1 Conv. % (e.e.%) | Sub 2 Conv. % (e.e.%) | Sub3 Conv. % (e.e.%) |
|---|---|---|---|---|
| 1 | Phosphate 0.1 M pH 8 | 56 (20, R) (a) | 30 (36, R) | 92 (42, R) |
| 2 | MOPS 1.2 M pH 7.8 | 34 (rac) | 40 (46, R) | 64 (rac) |
| 3 | MES 1.2 M pH 7 | 82 (4, S) | 30 (9, R) | 60 (4, R) |
| 4 | MES 1.2 M pH 6 | 40 (4, S) | 67 (12, R) | 25 (rac) |
| 5 | Acetate 0.1 M pH 5 | 34 (3, S) | 20 (21, R) | 30 (rac) |
| 6 | MES 1.2 M pH 5 | 75 (5, S) | 35 (61, R) | 20 (30, S) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Facchetti, G.; Pellegrino, S.; Bucci, R.; Nava, D.; Gandolfi, R.; Christodoulou, M.S.; Rimoldi, I. Vancomycin-Iridium (III) Interaction: An Unexplored Route for Enantioselective Imine Reduction. Molecules 2019, 24, 2771. https://doi.org/10.3390/molecules24152771
Facchetti G, Pellegrino S, Bucci R, Nava D, Gandolfi R, Christodoulou MS, Rimoldi I. Vancomycin-Iridium (III) Interaction: An Unexplored Route for Enantioselective Imine Reduction. Molecules. 2019; 24(15):2771. https://doi.org/10.3390/molecules24152771
Chicago/Turabian StyleFacchetti, Giorgio, Sara Pellegrino, Raffaella Bucci, Donatella Nava, Raffaella Gandolfi, Michael S. Christodoulou, and Isabella Rimoldi. 2019. "Vancomycin-Iridium (III) Interaction: An Unexplored Route for Enantioselective Imine Reduction" Molecules 24, no. 15: 2771. https://doi.org/10.3390/molecules24152771