3.2. Final Products Characterization
The purification method is specified here only when altered from the generally used method described in
Supplementary information.
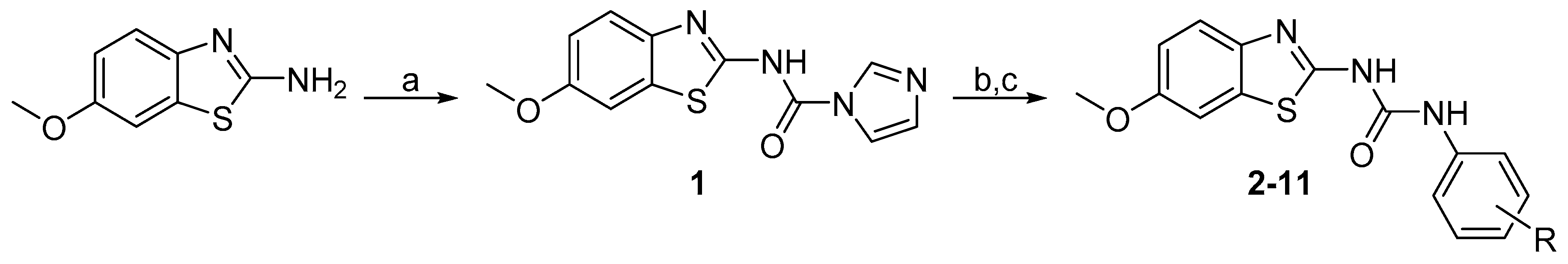
1-(4-Hydroxy-3-methylphenyl)-3-(6-methoxybenzo[d]thiazol-2-yl)urea (2) Yield 85%; mp: 262–263 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.55 (br s, 1H), 9.09 (s, 1H), 8.77 (s, 1H), 7.54 (d, J = 8.8 Hz, 1H), 7.50 (d, J = 2.6 Hz, 1H), 7.18 (d, J = 2.6 Hz, 1H), 7.10 (dd, J = 8.5, 2.7 Hz, 1H), 6.97 (dd, J = 8.8, 2.6 Hz, 1H), 6.73 (d, J = 8.5 Hz, 1H), 3.79 (s, 3H), 2.12 (s, 3H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 157.57, 155.61, 151.75, 151.47, 142.84, 132.53, 129.56, 124.11, 122.24, 120.13, 118.18, 114.64, 114.29, 104.87, 55.60, 16.12; HRMS (ESI) calcd for C16H16N3O3S [M + H]+ 330.09069, found 330.09039.
1-(3-(Tert-butyl)-4-hydroxyphenyl)-3-(6-methoxybenzo[d]thiazol-2-yl)urea (3) Yield 73%; mp: 259–260 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.47 (br s, 1H), 9.18 (br s, 1H), 8.79 (s, 1H), 7.54 (d, J = 8.8 Hz, 1H), 7.49 (d, J = 2.6 Hz, 1H), 7.19 (d, J = 2.6 Hz, 1H), 7.17 (dd, J = 8.4, 2.6 Hz, 1H), 6.97 (dd, J = 8.8, 2.6 Hz, 1H), 6.73 (d, J = 8.4 Hz, 1H), 3.79 (s, 3H), 1.35 (s, 9H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 157.50, 155.61, 151.91, 151.84, 142.82, 135.53, 132.60, 129.43, 120.20, 118.58, 118.38, 116.13, 114.28, 104.90, 55.62, 34.35, 29.25; HRMS (ESI) calcd for C19H22N3O3S [M + H]+ 372.13764, found 372.13730.
1-(3-Cyano-4-hydroxyphenyl)-3-(6-methoxybenzo[d]thiazol-2-yl)urea (4) Yield 98%; mp: 277–279 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.86 (s, 1H), 9.37 (s, 1H), 7.76 (d, J = 2.7 Hz, 1H), 7.56 – 7.52 (m, 2H), 7.51 (d, J = 2.6 Hz, 1H), 7.01 (d, J = 9.0 Hz, 1H), 6.98 (dd, J = 8.8, 2.6 Hz, 1H), 3.79 (s, 3H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 157.84, 156.05, 155.71, 152.44, 141.78, 132.24, 130.54, 126.52, 122.87, 119.79, 116.80, 116.73, 114.40, 105.00, 98.49, 55.61; HRMS (ESI) calcd for C16H12N4O3S [M + H]+ 341.07029, found 341.07016.
1-(3-Bromo-4-hydroxyphenyl)-3-(6-methoxybenzo[d]thiazol-2-yl)urea (5) Yield 82%; mp: 246–247 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.71 (br s, 1H), 9.99 (s, 1H), 8.97 (s, 1H), 7.75 (s, 1H), 7.54 (d, J = 8.4 Hz, 1H), 7.50 (s, 1H), 7.22 (d, J = 8.6 Hz, 1H), 6.97 (d, J = 8.5 Hz, 1H), 6.92 (d, J = 8.6 Hz, 1H), 3.79 (s, 3H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 157.84, 155.67, 152.21, 149.97, 142.25, 132.33, 131.04, 123.65, 120.14, 119.87, 116.28, 114.37, 108.86, 104.95, 55.61; HRMS (ESI) calcd for C15H13BrN3O3S [M + H]+ 393.98555, found 393.98489.
1-(4-Hydroxy-3-iodophenyl)-3-(6-methoxybenzo[d]thiazol-2-yl)urea (6) Yield 85%; mp: 241–242 °C; 1H-NMR (300 MHz, DMSO-d6): δ (ppm) 10.07 (br s, 1H), 9.05 (s, 1H), 7.90 (d, J = 2.6 Hz, 1H), 7.54 (d, J = 8.8 Hz, 1H), 7.51 (d, J = 2.6 Hz, 1H), 7.25 (dd, J = 8.7, 2.6 Hz, 1H), 6.97 (dd, J = 8.8, 2.6 Hz, 1H), 6.85 (d, J = 8.7 Hz, 1H), 3.79 (s, 3H); 13C-NMR (75 MHz, DMSO-d6): δ (ppm) 157.72, 155.67, 152.67, 152.10, 142.15, 132.35, 131.29, 129.42, 120.99, 119.93, 114.75, 114.38, 104.93, 84.10, 55.62; HRMS (ESI) calcd for C15H13IN3O3S [M + H]+ 441.97168, found 441.97049.
1-(3-Amino-4-hydroxyphenyl)-3-(6-methoxybenzo[d]thiazol-2-yl)urea (7) Yield 53%; mp: 180–181 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.41 (br s, 1H), 8.76 (br s, 1H), 8.65 (s, 1H), 7.53 (d, J = 8.8 Hz, 1H), 7.49 (d, J = 2.6 Hz, 1H), 6.96 (dd, J = 8.8, 2.6 Hz, 1H), 6.82 (d, J = 2.6 Hz, 1H), 6.57 (d, J = 8.3 Hz, 1H), 6.47 (dd, J = 8.3, 2.5 Hz, 1H), 4.61 (s, 2H), 3.79 (s, 3H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 157.45, 155.57, 151.34, 143.00, 140.11, 136.90, 132.57, 130.42, 122.10, 120.22, 114.24, 107.26, 106.17, 104.84, 55.58; HRMS (ESI) calcd for C15H15N4O3S [M + H]+ 331.08594, found 331.08527.
1-(6-Hydroxypyridin-3-yl)-3-(6-methoxybenzo[d]thiazol-2-yl)urea (8) Yield 82%; mp: 272–273 °C; 1H-NMR (300 MHz, DMSO-d6): δ (ppm) 11.38 (br s, 1H), 9.08 (br s, 1H), 7.64 (s, 1H), 7.59 (d, J = 9.1 Hz, 1H), 7.53 – 7.39 (m, 2H), 6.97 (dd, J = 9.2, 2.6 Hz, 1H), 6.36 (d, J = 10.1 Hz, 1H), 3.79 (s, 3H); 13C-NMR (75 MHz, DMSO-d6): δ (ppm) 160.85, 158.50, 155.83, 153.21, 142.19, 138.04, 132.42, 127.92, 120.11, 119.28, 119.17, 114.49, 105.11, 55.78; HRMS (ESI) calcd for C14H13N4O3S [M + H]+ 317.07029, found 317.07004.
1-(4-Aminophenyl)-3-(6-methoxybenzo[d]thiazol-2-yl)urea (9) Yield 93%; mp: 304–306 °C (decomp); 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.46 (br s, 1H), 8.63 (s, 1H), 7.54 (d, J = 8.8 Hz, 1H), 7.50 (d, J = 2.6 Hz, 1H), 7.15 – 7.10 (m, 2H), 6.96 (dd, J = 8.8, 2.6 Hz, 1H), 6.57 – 6.52 (m, 2H), 4.90 (s, 2H), 3.79 (s, 3H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 157.58, 155.56, 151.75, 144.96, 142.74, 132.56, 127.01, 121.23, 120.12, 114.23, 114.05, 104.87, 55.58; HRMS (ESI) calcd for C15H15N4O2S [M + H]+ 315.09102, found 315.09058.
1-(4-Amino-3-chlorophenyl)-3-(6-methoxybenzo[d]thiazol-2-yl)urea (10) Yield 78%y mp: 310-311 °C (decomp); 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.60 (br s, 1H), 8.82 (s, 1H), 7.54 (d, J = 8.7 Hz, 1H), 7.50 (d, J = 2.6 Hz, 1H), 7.47 (d, J = 2.4 Hz, 1H), 7.06 (dd, J = 8.6, 2.4 Hz, 1H), 6.97 (dd, J = 8.8, 2.6 Hz, 1H), 6.78 (d, J = 8.6 Hz, 1H), 5.14 (s, 2H), 3.79 (s, 3H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 157.69, 155.61, 151.90, 140.78, 132.42, 128.05, 120.50, 120.23, 119.97, 116.77, 115.48, 114.29, 104.91, 55.59; HRMS (ESI) calcd for C15H14ClN4O2S [M + H]+ 349.05205, found 349.05154.
1-(3-Chloro-4-(hydroxymethyl)phenyl)-3-(6-methoxybenzo[d]thiazol-2-yl)urea (11) Yield 92%; mp: 229–230 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.84 (br s, 1H), 9.26 (s, 1H), 7.72 (d, J = 2.2 Hz, 1H), 7.55 (d, J = 8.7 Hz, 1H), 7.51 (d, J = 2.6 Hz, 1H), 7.47 (d, J = 8.4 Hz, 1H), 7.38 (dd, J = 8.4 Hz, 2.1 Hz, 1H), 6.98 (dd, J = 8.8, 2.6 Hz, 1H), 5.29 (t, J = 5.6 Hz, 1H), 4.53 (d, J = 5.2 Hz, 2H), 3.80 (s, 3H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 157.75, 155.74, 152.27, 138.45, 133.63, 132.18, 131.19, 128.67, 127.18, 119.85, 118.56, 117.35, 114.44, 105.01, 59.99, 55.61; HRMS (ESI) calcd for C16H15ClN3O3S [M + H]+ 364.05172, found 364.05103.
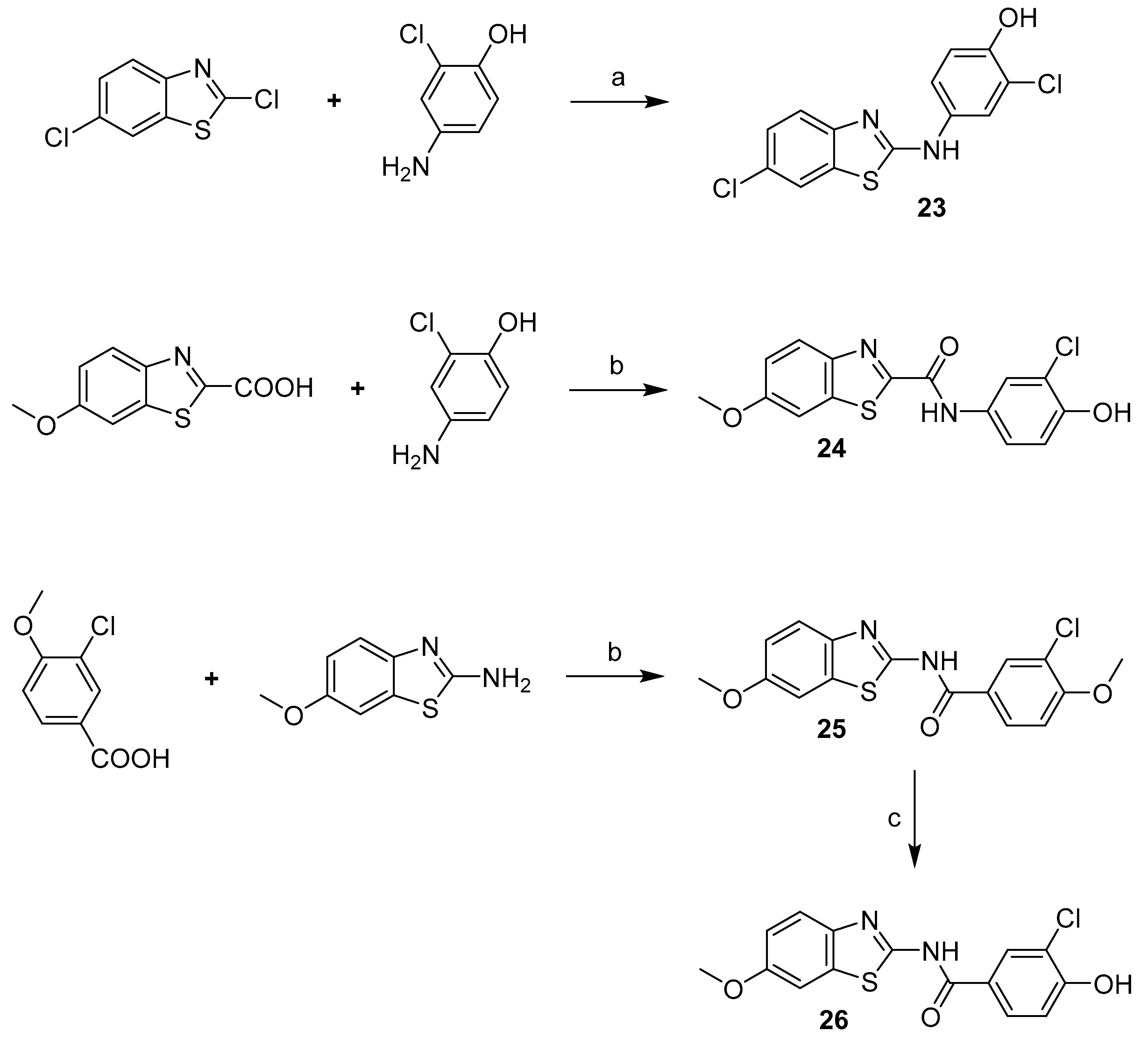
2-Chloro-4-((6-chlorobenzo[d]thiazol-2-yl)amino)phenol (23) Yield 30%; mp: 213–214.5 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.49 (br s, 1H), 9.92 (br s, 1H), 7.94 – 7.85 (m, 2H), 7.54 (d, J = 8.6 Hz, 1H), 7.40 (dd, J = 8.8, 2.6 Hz, 1H), 7.31 (dd, J = 8.6, 2.2 Hz, 1H), 6.98 (d, J = 8.8 Hz, 1H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 162.53, 150.83, 148.56, 132.94, 131.53, 126.01, 125.88, 120.74, 119.86, 119.74, 119.43, 118.46, 116.87; HRMS (ESI) calcd for C13H8Cl2N2OS [M + H]+ 310.98072, found 310.98016.
3-Chloro-4-hydroxy-N-(6-methoxybenzo[d]thiazol-2-yl)benzamide (24) Yield 69%; mp: 301.5–302.5 °C; 1H-NMR (300 MHz, DMSO-d6): δ (ppm) 12.51 (br s, 1H), 11.25 (br s, 1H), 8.21 (s, 1H), 7.97 (d, J = 8.6 Hz, 1H), 7.66 (d, J = 8.9 Hz, 1H), 7.59 (s, 1H), 7.17 – 6.96 (m, 2H), 3.82 (s, 3H); 13C-NMR (75 MHz, DMSO-d6): δ (ppm) 164.08, 157.27, 156.85, 156.21, 142.57, 132.85, 130.34, 128.93, 123.50, 120.96, 119.89, 116.34, 115.00, 104.66, 55.64; HRMS (ESI) calcd for C15H11ClN2O3S [M + H]+ 335.02517, found 335.02466.
N-(3-Chloro-4-hydroxyphenyl)-6-methoxybenzo[d]thiazole-2-carboxamide (26) Yield 58%; mp: 260–261.5 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.94 (br s, 1H), 10.09 (br s, 1H), 8.06 (d, J = 8.4 Hz, 1H), 7.94 (s, 1H), 7.80 (s, 1H), 7.65 (d, J = 7.7 Hz, 1H), 7.23 (d, J = 8.1 Hz, 1H), 6.97 (d, J = 8.2 Hz, 1H), 3.88 (s, 3H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 161.76, 158.62, 157.88, 149.87, 147.05, 138.22, 130.34, 124.71, 122.22, 120.81, 119.09, 117.30, 116.36, 104.79, 55.84; HRMS (ESI) calcd for C15H11ClN2O3S [M + H]+ 335.02517, found 335.02466.
1-(3-Chloro-4-hydroxybenzyl)-3-(6-methoxybenzo[d]thiazol-2-yl)urea (28)
The crude product was purified using column chromatography.
Yield 20%; mp: 249–251 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.60 (br s, 1H), 10.08 (br s, 1H), 7.51 (d, J = 8.8 Hz, 1H), 7.48 (d, J = 2.6 Hz, 1H), 7.29 (d, J = 2.1 Hz, 1H), 7.13 (t, J = 5.3 Hz, 1H), 7.10 (dd, J = 8.3, 2.1 Hz, 1H), 6.98 – 6.90 (m, 2H), 4.25 (d, J = 5.9 Hz, 2H), 3.78 (s, 3H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 157.80, 155.52, 153.84, 152.01, 143.16, 132.59, 131.25, 128.80, 127.18, 120.19, 119.37, 116.54, 114.15, 104.81, 55.57, 42.01; HRMS (ESI) calcd for C16H14ClN3O3S [M + H]+ 364.05172, found 364.05115.
1-(3,4-Dihydroxybenzyl)-3-(6-methoxybenzo[d]thiazol-2-yl)urea (29)
After the reaction was completed (monitored by TLC), 1M aq. HCl was poured to the reaction mixture and the product was extracted to DCM. The organic layer was concentrated and the crude product was recrystallized from MeCN.
Yield 50%; mp: 141.5–142 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 7.51 (d, J = 8.8 Hz, 1H), 7.48 (d, J = 2.6 Hz, 1H), 7.15 (br s, 1H), 6.95 (dd, J = 8.8, 2.6 Hz, 1H), 6.71 (d, J = 2.0 Hz, 1H), 6.68 (d, J = 8.0 Hz, 1H), 6.56 (dd, J = 8.0, 2.0 Hz, 1H), 4.18 (d, J = 5.5 Hz, 2H), 3.78 (s, 3H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 158.51, 155.74, 153.64, 145.23, 144.41, 141.36, 131.95, 129.93, 119.61, 118.26, 115.49, 114.93, 114.45, 105.07, 55.65, 42.68; HRMS (ESI) calcd for C16H15N3O4S [M + H]+ 346.08560, found 346.08517.
Dimethyl ((4-fluorophenyl)((6-methoxybenzo[d]thiazol-2-yl)amino)methyl) phosphonate (34) (Valasani et al. 2013).
Yield 65%; mp: 173–174 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 9.48 (s, 1H), 8.76 (dd, J = 9.7, 2.9 Hz, 1H), 7.33 – 7.27 (m, 4H), 6.82 (dd, J = 8.7, 2.7 Hz, 1H), 6.75 (d, J = 8.4 Hz, 2H), 5.54 (dd, J = 20.9, 9.7 Hz, 1H), 3.72 (s, 3H), 3.64 (d, J = 10.5 Hz, 3H), 3.50 (d, J = 10.5 Hz, 3H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 163.62 (d, J = 10.0 Hz), 161.69 (dd, J = 244.1, 2.9 Hz), 154.67, 145.52, 132.10 (d, J = 2.9 Hz), 131.87, 130.02 (dd, J = 8.3, 5.5 Hz), 118.79, 115.18 (dd, J = 21.6, 1.7 Hz), 113.09, 105.58, 53.53 (d, J = 6.8 Hz), 53.25 (d, J = 6.8 Hz), 53.12 (d, J = 154.1 Hz); 31P NMR (202 MHz, DMSO-d6): δ (ppm) 23.56 (d, J = 4.5 Hz); HRMS (ESI) calcd for C17H19FN2O4PS [M + H]+ 397.07817, found 397.07755.
Dimethyl ((4-hydroxyphenyl)((6-methoxybenzo[d]thiazol-2-yl)amino)methyl) phosphonate (35) (Valasani et al. 2013). Yield 76%; mp: 217–218 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 9.48 (s, 1H), 8.76 (dd, J = 9.7, 2.9 Hz, 1H), 7.33 – 7.28 (m, 4H), 6.82 (dd, J = 8.7, 2.7 Hz, 1H), 6.75 (d, J = 8.4 Hz, 2H), 5.54 (dd, J = 20.9, 9.7 Hz, 1H), 3.72 (s, 3H), 3.64 (d, J = 10.5 Hz, 3H), 3.50 (d, J = 10.5 Hz, 3H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 163.70 (d, J = 10.0 Hz), 157.06 (d, J = 2.4 Hz), 154.57, 145.64, 131.82, 129.31 (d, J = 5.7 Hz), 125.77, 118.68, 115.08, 113.01, 105.57, 55.53, 53.33 (d, J = 6.9 Hz), 53.28 (d, J = 155.2 Hz), 53.14 (d, J = 6.9 Hz); 31P NMR (202 MHz, DMSO-d6): δ (ppm) 24.25; HRMS (ESI) calcd for C17H20N2O5PS [M + H]+ 395.08251, found 395.08215.
Methyl 5-((dimethoxyphosphoryl)((6-methoxybenzo[d]thiazol-2-yl)amino)methyl)-2- hydroxybenzoate (36) (Valasani et al. 2013) Yield 73%; mp: 180–181 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.53 (s, 1H), 8.90 (dd, J = 9.6, 3.5 Hz, 1H), 7.95 (t, J = 2.3 Hz, 1H), 7.65 (dt, J = 8.7, 2.1 Hz, 1H), 7.32 (d, J = 2.7 Hz, 1H), 7.30 (d, J = 8.7 Hz, 1H), 7.01 (d, J = 8.6 Hz, 1H), 6.82 (dd, J = 8.8, 2.7 Hz, 1H), 5.63 (dd, J = 21.3, 9.4 Hz, 1H), 3.91 (s, 3H), 3.72 (s, 3H), 3.67 (d, J = 10.6 Hz, 3H), 3.55 (d, J = 10.6 Hz, 3H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 168.86, 163.61 (d, J = 10.1 Hz), 159.53 (d, J = 2.0 Hz), 154.65, 145.52, 135.35 (d, J = 5.1 Hz), 131.88, 129.33 (d, J = 6.0 Hz), 126.83, 118.79, 117.49, 105.58, 113.07, 113.03 (d, J = 1.9 Hz), 55.53, 53.55 (d, J = 7.1 Hz), 53.26 (d, J = 6.8 Hz), 52.90 (d, J = 155.2 Hz), 52.53; 31P NMR (202 MHz, DMSO-d6): δ (ppm) 23.67; HRMS (ESI) calcd for C19H22N2O7PS [M + H]+ 453.08798, found 453.08701.
Dimethyl ((3-chloro-4-hydroxyphenyl)((6-methoxybenzo[d]thiazol-2-yl)amino)methyl) phosphonate (37) Yield 65%; mp: 198–199 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.28 (s, 1H), 8.77 (dd, J = 9.7, 3.1 Hz, 1H), 7.50 (t, J = 2.1 Hz, 1H), 7.32 (d, J = 2.7 Hz, 1H), 7.31 (d, J = 8.8 Hz, 1H), 7.27 (dt, J = 8.5, 2.1 Hz, 1H), 6.95 (d, J = 8.4 Hz, 1H), 6.82 (dd, J = 8.7, 2.6 Hz, 1H), 5.57 (dd, J = 21.0, 9.6 Hz, 1H), 3.73 (s, 3H), 3.66 (d, J = 10.6 Hz, 3H), 3.55 (d, J = 10.6 Hz, 3H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 163.76 (d, J = 10.1 Hz), 154.82, 152.90 (d, J = 2.4 Hz), 145.72, 132.01, 129.40 (d, J = 5.4 Hz), 128.12 (d, J = 5.9 Hz), 127.58, 119.66 (d, J = 2.2 Hz), 118.94, 116.52, 113.25, 105.76, 55.71, 53.66 (d, J = 6.8 Hz), 53.41 (d, J = 7.0 Hz), 52.95 (d, J = 155.3 Hz); 31P NMR (202 MHz, DMSO-d6): δ (ppm) 23.71; HRMS (ESI) calcd for C17H19ClN2O5PS [M + H]+ 429.04353, found 429.0425.
1-(Benzo[d]thiazol-2-yl)-3-(3-chloro-4-hydroxyphenyl)-1-methylurea (41) Yield 83%; mp: 171–172 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.02 (s, 1H), 9.47 (s, 1H), 7.94 – 7.84 (m, 1H), 7.74 (d, J = 7.9 Hz, 1H), 7.57 (d, J = 2.6 Hz, 1H), 7.45 – 7.35 (m, 1H), 7.31 (dd, J = 8.8, 2.6 Hz, 1H), 7.28 – 7.19 (m, 1H), 6.96 (d, J = 8.7 Hz, 1H), 3.75 (s, 3H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 161.45, 153.45, 149.66, 148.45, 132.76, 130.50, 125.80, 123.42, 123.09, 121.95, 121.12, 120.29, 119.01, 116.29, 34.55 (d, J = 3.0 Hz); HRMS (ESI) calcd for C15H12ClN3O2S [M + H]+ 334.04115, found 334.04080.
3-(Benzo[d]thiazol-2-yl)-1-(3-chloro-4-hydroxyphenyl)-1-methylurea (45)
The crude product was dissolved in Et2O and filtered. To the filtrate was added PE and the solution was left to crystallize in a freezer. Filtration gave the desired pure product.
Yield 53%; mp: 139–141 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.28 (br s, 1H), 7.81 (d, J = 7.5 Hz, 1H), 7.45 (br s, 1H), 7.38 – 7.30 (m, 2H), 7.19 (t, J = 7.9 Hz, 1H), 7.11 (dd, J = 8.6, 2.5 Hz, 1H), 6.99 (d, J = 8.6 Hz, 1H), 3.26 (s, 3H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 151.90, 135.11, 128.70, 126.89, 125.94, 122.65, 121.62, 119.46, 116.66, 37.93; HRMS (ESI) calcd for C15H12ClN3O2S [M + H]+ 334.0412, found 334.0414.
1-(3-Chloro-4-hydroxyphenyl)-3-(6-methoxybenzo[d]thiazol-2-yl)-1-methylurea (46)
The crude product was purified using column chromatography.
Yield 50%; mp: 220 °C decomp.; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.28 (br s, 1H), 7.45 (d, J = 2.2 Hz, 1H), 7.38 (d, J = 8.1 Hz, 1H), 7.36 (d, J = 2.5 Hz, 1H), 7.11 (dd, J = 8.6, 2.5 Hz, 1H), 6.98 (d, J = 8.6 Hz, 1H), 6.94 (dd, J = 8.8, 2.6 Hz, 1H), 3.77 (s, 3H), 3.25 (s, 3H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 155.57, 151.97, 134.94, 131.53, 128.78, 126.95, 119.52, 118.40, 116.70, 114.18, 105.12, 55.60, 37.94; HRMS (ESI) calcd for C16H14ClN3O3S [M + H]+ 364.0517, found 364.0530.
1-(Benzo[d]thiazol-2-yl)-3-(3-chloro-4-hydroxyphenyl)-1,3-dimethylurea (49) Yield 65%; mp: 227–228.5 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.09 (s, 1H), 7.74 (d, J = 7.7 Hz, 1H), 7.47 – 7.37 (m, 2H), 7.36 (d, J = 2.4 Hz, 1H), 7.23 (t, J = 7.9 Hz, 1H), 7.13 (dd, J = 8.6, 2.2 Hz, 1H), 6.95 (d, J = 8.7 Hz, 1H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 165.20, 160.92, 150.53, 137.50, 136.58, 127.60, 126.56, 125.93, 125.38, 123.01, 122.40, 118.67, 115.86, 111.35, 37.16, 31.51; HRMS (ESI) calcd for C16H14ClN3O2S [M + H]+ 348.05680, found 348.05661.
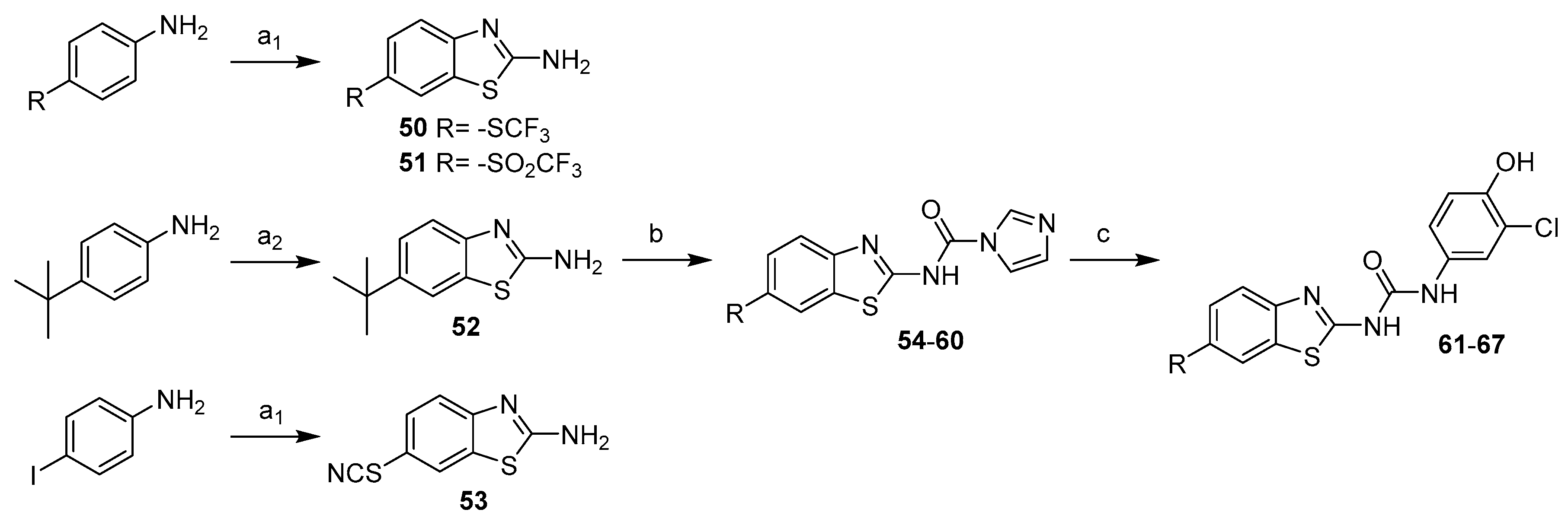
1-(3-Chloro-4-hydroxyphenyl)-3-(6-isopropylbenzo[d]thiazol-2-yl)urea (61) Yield 43%; mp: 246–248 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.83 (br s, 1H), 9.91 (br s, 1H), 9.02 (s, 1H), 7.74 (s, 1H), 7.61 (d, J = 2.4 Hz, 1H), 7.54 (d, J = 8.2 Hz, 1H), 7.26 (dd, J = 8.3, 1.5 Hz, 1H), 7.19 (dd, J = 8.7, 2.4 Hz, 1H), 6.93 (d, J = 8.7 Hz, 1H), 6.73 (d, J = 8.7 Hz, 1H), 2.97 (sept, J = 6.9 Hz, 1H), 1.23 (d, J = 6.9 Hz, 6H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 159.33, 152.47, 148.89, 146.08, 143.46, 131.02, 130.84, 124.67, 120.74, 119.41, 119.33, 118.68, 116.64, 33.41, 24.17; HRMS (ESI) calcd for C17H16ClN3O2S [M + H]+ 362.0725, found 362.0721.
1-(6-(Tert-butyl)benzo[d]thiazol-2-yl)-3-(3-chloro-4-hydroxyphenyl)urea (62) Yield 66%; mp: 238–240 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 9.75 (s, 1H), 7.90 (d, J = 1.9 Hz, 1H), 7.60 (d, J = 2.6 Hz, 1H), 7.56 (d, J = 8.5 Hz, 1H), 7.43 (dd, J = 8.5, 2.0 Hz, 1H), 7.18 (dd, J = 8.8, 2.6 Hz, 1H), 6.95 (d, J = 8.7 Hz, 1H), 1.32 (s, 9H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 159.43, 152.17, 148.89, 145.88, 145.02, 130.87, 130.77, 123.74, 120.40, 119.36, 119.08, 118.41, 117.78, 116.74, 34.62, 31.45; HRMS (ESI) calcd for C18H18ClN3O2S [M + H]+ 376.0881, found 376.0882.
1-(3-Chloro-4-hydroxyphenyl)-3-(6-ethoxybenzo[d]thiazol-2-yl)urea (63) Yield 90%; mp: 255–257 °C; 1H-NMR (300 MHz, DMSO-d6): δ (ppm) 10.72 (br s, 1H), 9.92 (s, 1H), 9.00 (s, 1H), 7.60 (s, 1H), 7.57–7.36 (m, 2H), 7.18 (d, J = 7.9 Hz, 1H), 7.04 – 6.84 (m, 2H), 4.04 (q, J = 6.9 Hz, 2H), 1.33 (t, J = 7.0 Hz, 3H); 13C-NMR (75 MHz, DMSO-d6): δ (ppm) 157.76, 154.90, 152.19, 148.90, 141.98, 132.33, 130.82, 120.74, 119.89, 119.41, 119.34, 116.67, 114.76, 105.59, 63.59, 14.75; HRMS (ESI) calcd for C16H14ClN3O3S [M + H]+ 364.0517, found 364.0521.
1-(3-Chloro-4-hydroxyphenyl)-3-(6-((trifluoromethyl)thio)benzo[d]thiazol-2-yl)urea (64) Yield 24%; mp: 256–258 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 11.04 (br s, 1H), 9.95 (br s, 1H), 9.03 (s, 1H), 8.37 (d, J = 1.8 Hz, 1H), 7.75 (d, J = 8.4 Hz, 1H), 7.67 (dd, J = 8.4, 1.9 Hz, 1H), 7.59 (d, J = 2.6 Hz, 1H), 7.19 (dd, J = 8.8, 2.6 Hz, 1H), 6.95 (d, J = 8.7 Hz, 1H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 162.27, 152.10, 150.64, 149.08, 134.08, 132.73, 130.61, 130.28, 129.69 (q, J = 308.1 Hz), 120.65, 120.34, 119.38, 119.33, 116.74, 115.69 (q, J = 2.1 Hz); HRMS (ESI) calcd for C15H9ClF3N3O2S2 [M + H]+ 419.9850, found 419.9873.
1-(3-Chloro-4-hydroxyphenyl)-3-(6-thiocyanatobenzo[d]thiazol-2-yl)urea (65) Yield 80%; mp: 251–253 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 11.04 (br s, 1H), 9.97 (br s, 1H), 9.13 (br s, 1H), 8.32 (d, J = 2.0 Hz, 1H), 7.74 (d, J = 8.5 Hz, 1H), 7.64 (dd, J = 8.5, 2.0 Hz, 1H), 7.60 (d, J = 2.6 Hz, 1H), 7.19 (dd, J = 8.8, 2.6 Hz, 1H), 6.94 (d, J = 8.7 Hz, 1H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 161.91, 152.15, 149.47, 149.14, 133.11, 130.46, 129.54, 125.55, 121.00, 120.73, 119.66, 119.35, 116.75, 116.66, 112.20; HRMS (ESI) calcd for C15H9ClN4O2S2 [M + H]+ 376.9928, found 376.9939.
1-(3-Chloro-4-hydroxyphenyl)-3-(6-(methylsulfonyl)benzo[d]thiazol-2-yl)urea (66) Yield 78%; mp: 293–295 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 11.16 (br s, 1H), 9.94 (br s, 1H), 9.25 (s, 1H), 8.55 (d, J = 1.7 Hz, 1H), 7.89 (dd, J = 8.5, 1.9 Hz, 1H), 7.82 (d, J = 8.5 Hz, 1H), 7.61 (d, J = 2.6 Hz, 1H), 7.20 (dd, J = 8.8, 2.6 Hz, 1H), 6.95 (d, J = 8.7 Hz, 1H), 3.23 (s, 3H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 163.39, 152.08, 149.15, 134.69, 131.79, 130.45, 124.81, 121.71, 120.89, 119.56, 119.36, 116.68, 44.09; HRMS (ESI) calcd for C15H12ClN3O4S2 [M + H]+ 398.0031, found 398.0048.
1-(3-Chloro-4-hydroxyphenyl)-3-(6-((trifluoromethyl)sulfonyl)benzo[d]thiazol-2-yl)urea (67) Yield 94%; mp: 267–269 °C; 1H-NMR (300 MHz, DMSO-d6): δ (ppm) 11.40 (br s, 1H), 10.01 (s, 1H), 9.10 (br s, 1H), 8.89 (s, 1H), 8.02 (dd, J = 8.6, 1.9 Hz, 1H), 7.95 (d, J = 8.4 Hz, 1H), 7.60 (d, J = 2.5 Hz, 1H), 7.21 (dd, J = 8.7, 2.3 Hz, 1H), 6.95 (d, J = 8.8 Hz, 1H); 13C-NMR (75 MHz, DMSO-d6): δ (ppm) 166.05, 155.92, 149.35, 133.47, 130.23, 128.07, 126.29, 121.75, 121.17, 119.85, 119.36, 117.44, 116.67, 40.35, 40.08, 39.80, 39.52, 39.24, 38.96, 38.69; HRMS (ESI) calcd for C15H9ClF3N3O4S2 [M + H]+ 451.9748, found 451.9749.
1-(3-Chloro-4-hydroxyphenyl)-3-(4,5,6,7-tetrahydrobenzo[d]thiazol-2-yl)urea (71) Yield 58%; mp: 258–260 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 9.91 (br s, 1H), 7.55 (d, J = 2.4 Hz, 1H), 7.14 (dd, J = 8.7, 2.4 Hz, 1H), 6.94 (d, J = 8.7 Hz, 1H), 2.59 (s, 2H), 2.54 (s, 2H), 1.75 (s, 4H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 158.38, 151.01, 149.02, 138.46, 130.54, 120.47, 120.31, 119.38, 119.00, 116.77, 24.13, 22.46, 22.07, 21.82; HRMS (ESI) calcd for C14H14ClN3O2S [M + H]+ 324.05680, found 324.05634.
1-(3-Chloro-4-hydroxyphenyl)-3-(2,3-dihydro-1H-inden-2-yl)urea (72) Yield 21%; mp: 203–205 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 9.59 (br s, 1H), 8.14 (br s, 1H), 7.52 (d, J = 2.6 Hz, 1H), 7.29 – 7.19 (m, 2H), 7.19 – 7.10 (m, 2H), 6.97 (dd, J = 8.7, 2.6 Hz, 1H), 6.83 (d, J = 8.7 Hz, 1H), 6.36 (d, J = 7.3 Hz, 1H), 4.48 – 4.31 (m, 1H), 3.25 – 3.09 (m, 2H), 2.84 – 2.67 (m, 2H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 155.00, 147.38, 141.23, 132.93, 126.40, 124.56, 119.34, 119.16, 117.90, 116.55, 50.77, 39.70; HRMS (ESI) calcd for C16H15ClN2O2 [M + H]+ 303.08948, found 303.08908.
1-(3-Chloro-4-hydroxyphenyl)-3-(4-(4-chlorophenyl)thiazol-2-yl)urea (73) Yield 71%; mp: 206–208 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 10.69 (s, 1H), 9.91 (s, 1H), 8.76 (s, 1H), 7.92 – 7.87 (m, 2H), 7.60 – 7.57 (m, 2H), 7.50 – 7.45 (m, 2H), 7.14 (dd, J = 8.7, 2.6 Hz, 1H), 6.93 (d, J = 8.7 Hz, 1H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 159.31, 151.59, 148.87, 147.41, 133.16, 132.07, 130.69, 128.67, 127.26, 120.67, 119.34, 116.66, 107.89; HRMS (ESI) calcd for C16H11Cl2N3O2S [M + H]+ 380.00218, found 380.00168.
1-(3-Chloro-4-hydroxyphenyl)-3-(thiazol-2-yl)urea (74) Yield 74%; mp: 220–222 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 9.83 (s, 1H), 9.81 (s, 1H), 7.56 (d, J = 2.6 Hz, 1H), 7.47 (dd, J = 3.9, 1.6 Hz, 1H), 7.20 (dd, J = 3.8, 1.5 Hz, 1H), 7.15 (dd, J = 8.8, 2.6 Hz, 1H), 6.95 (d, J = 8.7 Hz, 1H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 160.49, 151.25, 149.00, 133.59, 130.64, 120.41, 119.39, 119.08, 116.78, 113.06; HRMS (ESI) calcd for C10H8ClN3O2S [M + H]+ 270.0099, found 270.0099.
1-(3-Chloro-4-hydroxyphenyl)-3-(4-methoxyphenethyl)urea (75) Yield 21%; mp: 161.5–163.5 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 9.58 (br s, 1H), 8.29 (br s, 1H), 7.52 (d, J = 2.5 Hz, 1H), 7.14 (d, J = 8.5 Hz, 2H), 6.97 (dd, J = 8.7, 2.5 Hz, 1H), 6.89–6.84 (m, 2H), 6.82 (d, J = 8.7 Hz, 1H), 5.98 (t, J = 5.5 Hz, 1H), 3.72 (s, 3H), 3.33 – 3.19 (m, 2H), 2.66 (t, J = 7.2 Hz, 2H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 157.65, 155.20, 147.32, 133.06, 131.35, 129.58, 119.37, 119.14, 117.90, 116.54, 113.78, 54.97, 40.87, 34.96; HRMS (ESI) calcd for C16H17ClN2O3 [M + H]+ 321.10005, found 321.09967.
1-(Benzo[d]thiazol-6-yl)-3-(3-chloro-4-hydroxyphenyl)urea (76) Yield 24%; mp: 224–226 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 9.77 (s, 1H), 9.20 (s, 1H), 8.89 (s, 1H), 8.60 (s, 1H), 8.35 (d, J = 2.1 Hz, 1H), 7.97 (d, J = 8.8 Hz, 1H), 7.60 (d, J = 2.6 Hz, 1H), 7.47 (dd, J = 8.8, 2.2 Hz, 1H), 7.11 (dd, J = 8.7, 2.6 Hz, 1H), 6.90 (d, J = 8.7 Hz, 1H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 153.69, 152.69, 148.26, 148.17, 137.64, 134.46, 131.92, 122.92, 120.22, 119.26, 118.82, 118.11, 116.63, 110.26; HRMS (ESI) calcd for C14H10ClN3O2S [M + H]+ 320.0255, found 320.0264.
1,3-Bis(3-chloro-4-hydroxyphenyl)urea (78) Yield 94%; mp: 254.5–256.5 °C; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) 9.71 (br s, 2H), 8.43 (br s, 2H), 7.54 (d, J = 2.6 Hz, 2H), 7.06 (dd, J = 8.8, 2.6 Hz, 2H), 6.87 (d, J = 8.7 Hz, 2H); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) 152.77, 147.99, 132.14, 120.11, 119.22, 118.71, 116.59; HRMS (ESI) calcd for C13H10Cl2N2O3 [M + H]+ 313.01412, found 313.01361.

 ,
, 






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}























