
Inferring Drug-Related Diseases Based on Convolutional Neural Network and Gated Recurrent Unit

Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset
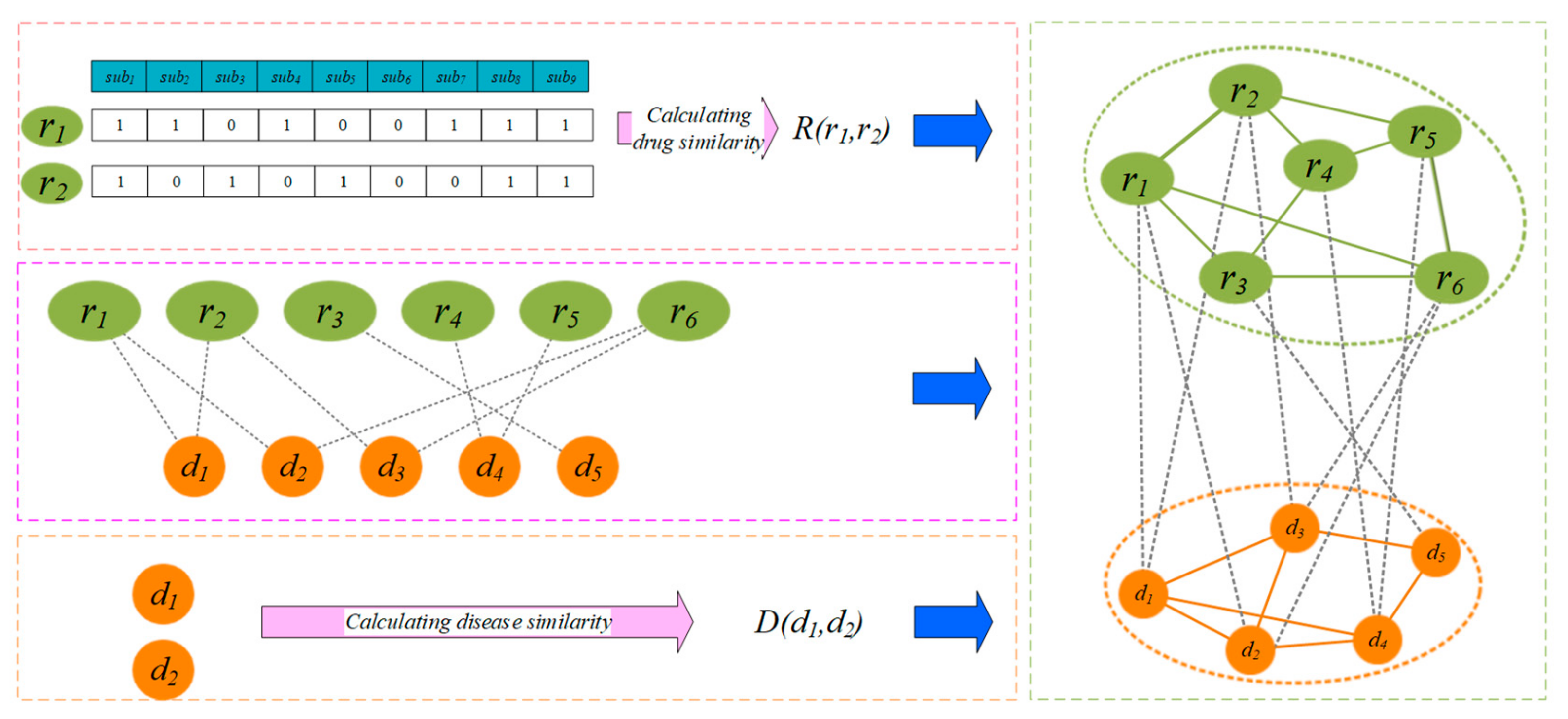
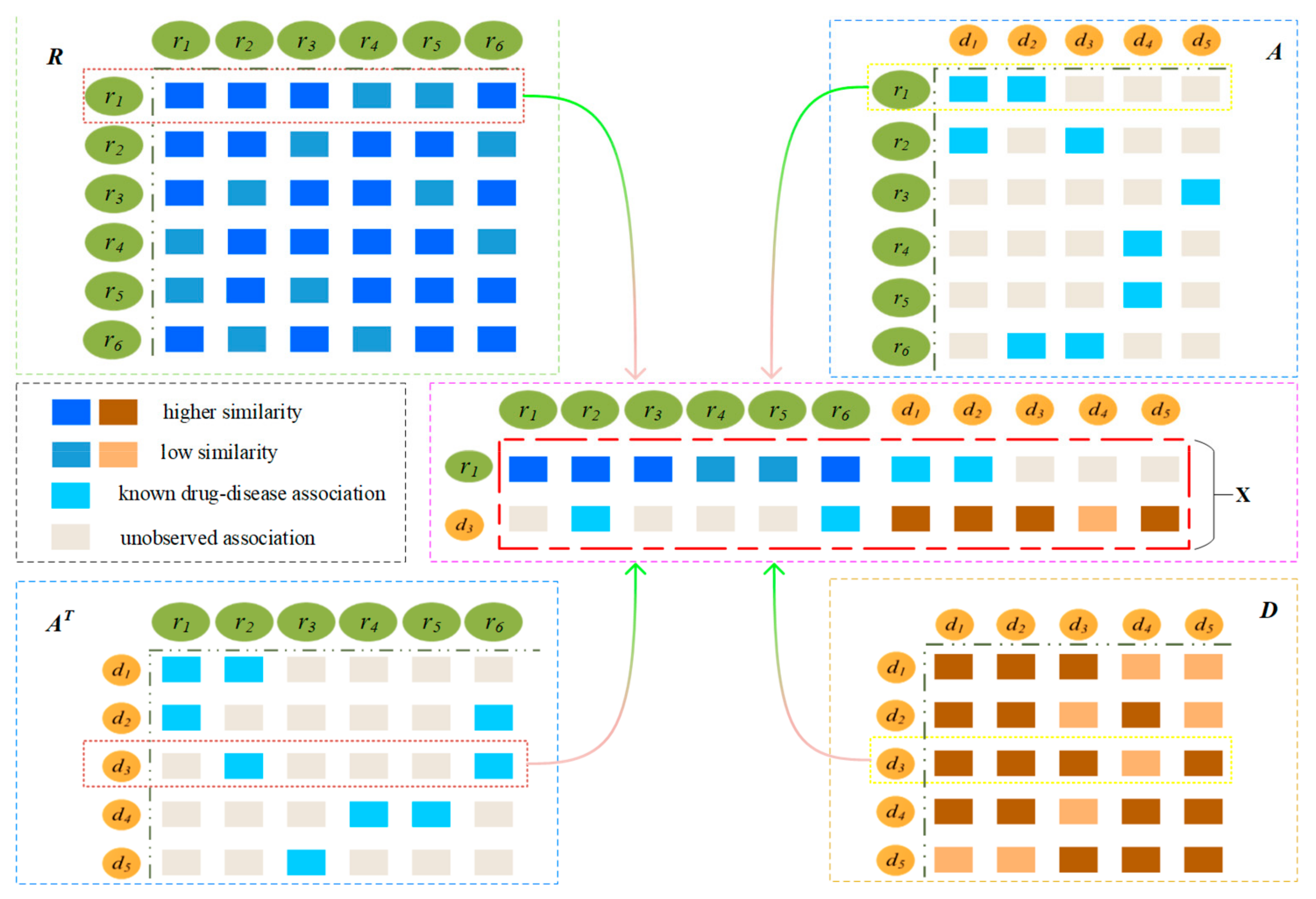
2.2. Construction of Drug-Disease Network
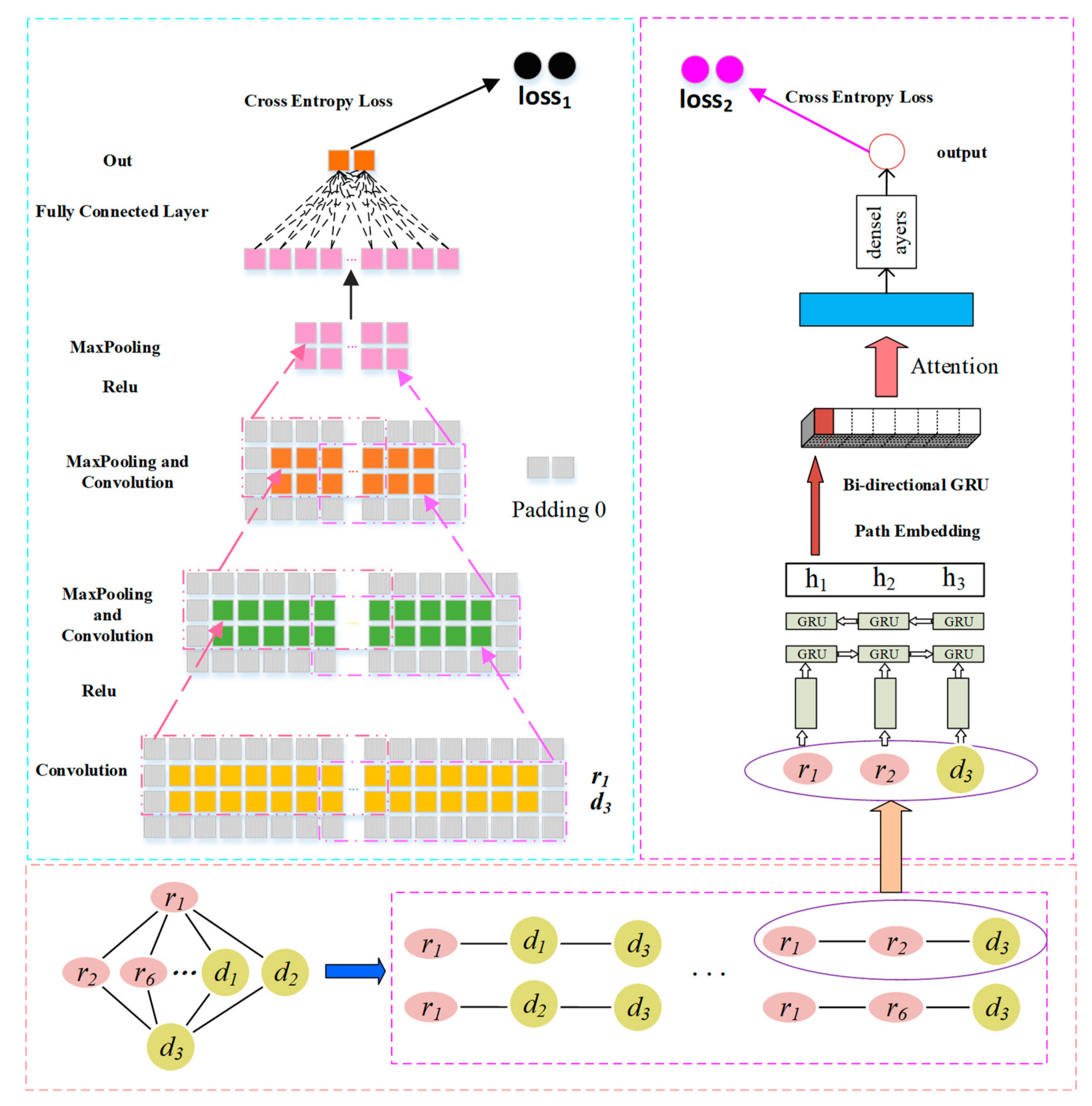
2.3. Prediction Model Based on CNN and GRU
2.3.1. Convolution Module on the Left
Convolutional Layer
Pooling Layer
2.3.2. GRU with Attention-Based Path Encoder on the Right
GRU-Based Sequence Encoder
GRU-Based Path Encoder
Path Attention
2.3.3. Combined Strategy
2.3.4. Reducing Overfitting
Dropout
3. Results and Discussion
3.1. Evaluation Metrics
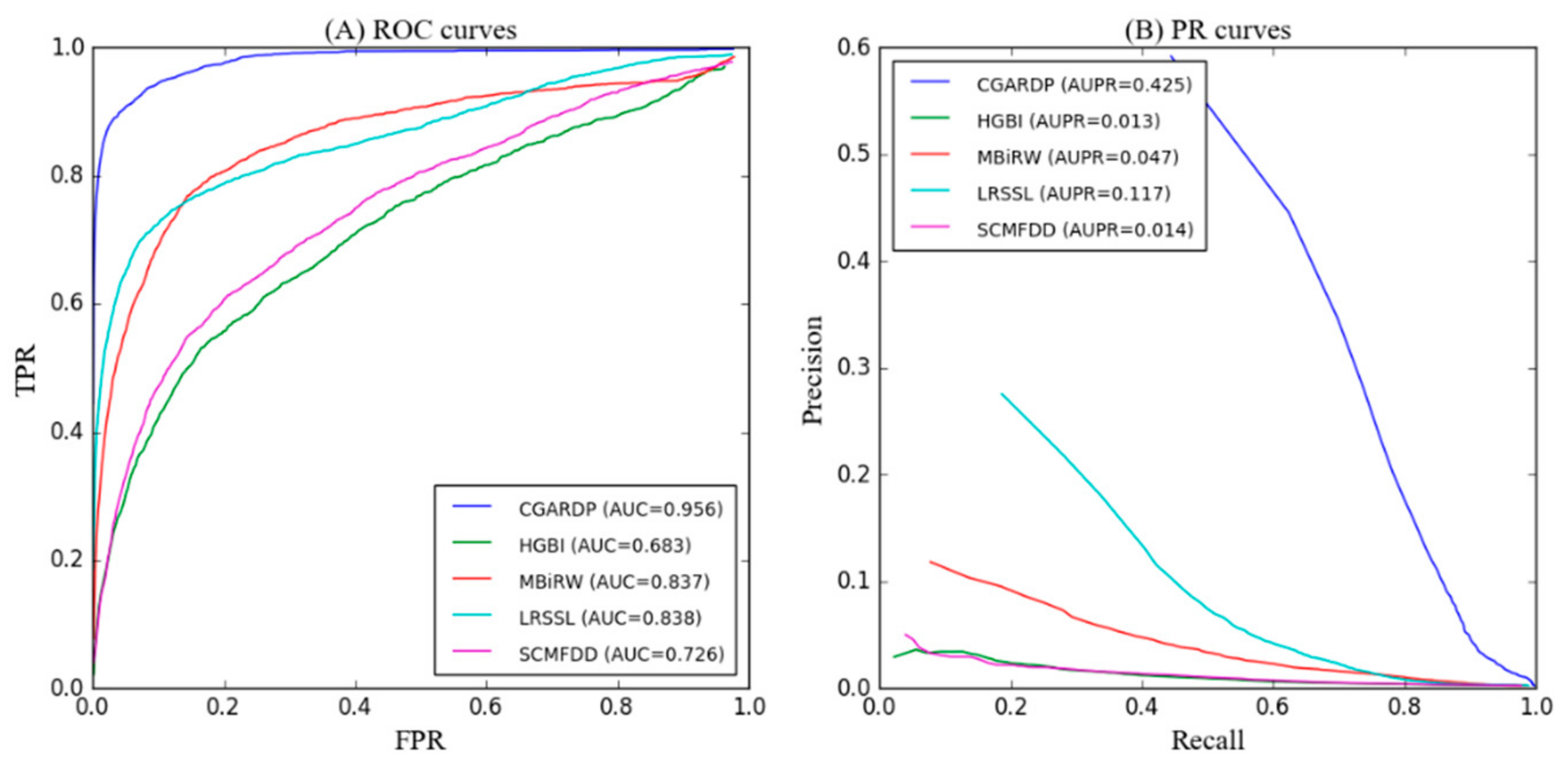
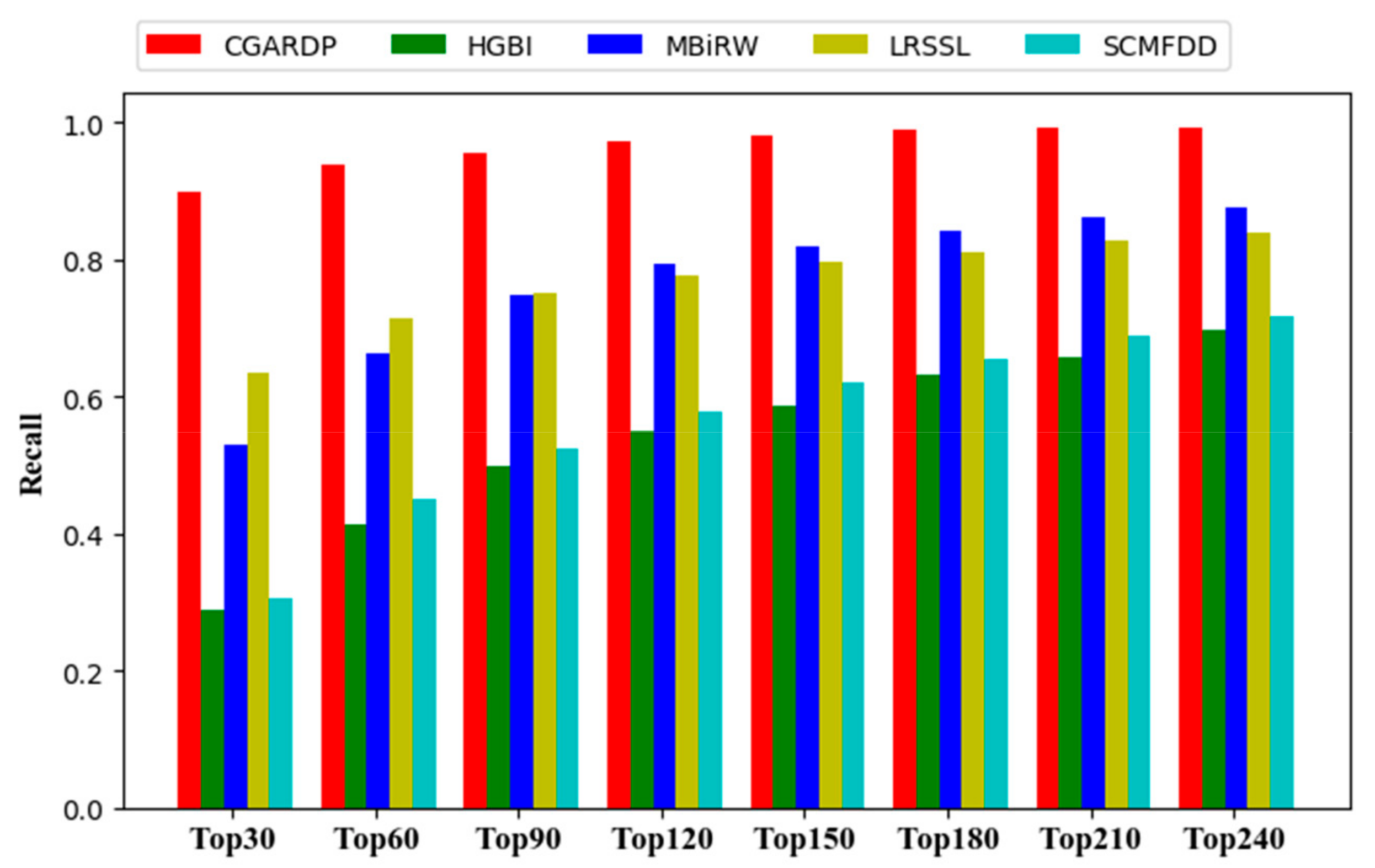
3.2. Comparison with Other Methods
3.3. Case Studies on Ciprofloxacin, Ceftriaxone, Ofloxacin, Ampicillin, and Levofloxacin
3.4. Prediction of Novel Drug–Disease Associations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Davis, J.; Goadrich, M. The relationship between Precision-Recall and ROC curves. In Proceedings of the 23rd international conference on Machine Learning, Pittsburgh, PA, USA, 25–29 June 2006; pp. 233–240. [Google Scholar]
- Mullard, A. 2014 FDA drug approvals. Nat. Rev. Drug Discov. 2015, 14, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Fashoyin-Aje, L.; Donoghue, M.; Chen, H.; He, K.; Veeraraghavan, J.; Goldberg, K.B.; Keegan, P.; McKee, A.E.; Pazdur, R. FDA Approval Summary: Pembrolizumab for Recurrent Locally Advanced or Metastatic Gastric or Gastroesophageal Junction Adenocarcinoma Expressing PD-L1. Oncologist 2019, 24, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.; Gagnon, J.P. Key factors in the rising cost of new drug discovery and development. Nat. Rev. Drug Discov. 2004, 3, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Ellis, P.; Tamimi, N. Drug Development: From Concept to Marketing! Nephron Clin. Pr. 2009, 113, c125–c131. [Google Scholar]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; Mcnamee, C. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41. [Google Scholar] [CrossRef] [PubMed]
- Alfedi, G.; Luffarelli, R.; Condò, I.; Pedini, G.; Mannucci, L.; Massaro, D.S.; Benini, M.; Toschi, N.; Alaimo, G.; Panarello, L.; et al. Drug repositioning screening identifies etravirine as a potential therapeutic for friedreich’s ataxia. Mov. Disord. 2019, 34, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Tobinick, E. The value of drug repositioning in the current pharmaceutical market. Drug News Perspect. 2009, 22, 119. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Suthram, S.; Dudley, J.T.; Chiang, A.P.; Chen, R.; Hastie, T.J.; Butte, A.J. Network-Based Elucidation of Human Disease Similarities Reveals Common Functional Modules Enriched for Pluripotent Drug Targets. PLoS Comput. Boil. 2010, 6, e1000662. [Google Scholar] [CrossRef]
- Chiang, A.P.; Butte, A.J.; Chiang, A.P.; Butte, A.J.; Chiang, A.; Butte, A. Systematic Evaluation of Drug–Disease Relationships to Identify Leads for Novel Drug Uses. Clin. Pharmacol. Ther. 2009, 86, 507–510. [Google Scholar] [CrossRef]
- Bamshad, M.J.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Zhang, Y.; Nguyen, H.G.; Koupenova, M.; Chauhan, A.K.; Makitalo, M.; Jones, M.R.; Hilaire, C.S.; Seldin, D.C.; Toselli, P.; et al. The A 2B adenosine receptor protects against inflammation and excessive vascular adhesion. J. Clin. Investig. 2006, 116, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.A.; Osterloh, I.H.; Grimminger, F. Sildenafil: From angina to erectile dysfunction to pulmonary hypertension and beyond. Nat. Rev. Drug Discov. 2006, 5, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Campillos, M.; Kuhn, M.; Gavin, A.-C.; Jensen, L.J.; Bork, P. Drug Target Identification Using Side-Effect Similarity. Science 2008, 321, 263–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sardana, D.; Zhu, C.; Zhang, M.; Gudivada, R.C.; Yang, L.; Jegga, A.G. Drug repositioning for orphan diseases. Briefings Bioinform. 2011, 12, 346–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.; Liu, C.; Jiang, J.; Lu, W.; Li, W.; Liu, G.; Zhou, W.-X.; Huang, J.; Tang, Y. Prediction of Drug-Target Interactions and Drug Repositioning via Network-Based Inference. PLoS Comput. Boil. 2012, 8, e1002503. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Li, S. A co-module approach for elucidating drug-disease associations and revealing their molecular basis. Bioinformatics 2012, 28, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, P.; Cao, N.; Hu, J.; Sorrentino, R. Exploring the associations between drug side-effects and therapeutic indications. J. Biomed. Inform. 2014, 51, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, A.; Stein, G.Y.; Ruppin, E.; Sharan, R. PREDICT: A method for inferring novel drug indications with application to personalized medicine. Mol. Syst. Biol. 2011, 7, 496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, F.; Hu, J. Towards drug repositioning: A unified computational framework for integrating multiple aspects of drug similarity and disease similarity. In Proceedings of the AMIA Annual Symposium Proceedings, Washington, DC, USA, 15–19 November 2014; pp. 1258–1267. [Google Scholar]
- Liang, X.; Zhang, P.; Yan, L.; Fu, Y.; Peng, F.; Qu, L.; Shao, M.; Chen, Y.; Chen, Z. LRSSL: Predict and interpret drug–disease associations based on data integration using sparse subspace learning. Bioinformatics 2017, 33, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Wang, J.; Li, M.; Peng, X.; Wu, F.-X.; Pan, Y. Drug repositioning based on comprehensive similarity measures and Bi-Random Walk algorithm. Bioinformatics 2016, 32, 2664–2671. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yue, X.; Lin, W.; Wu, W.; Liu, R.; Huang, F.; Liu, F. Predicting drug-disease associations by using similarity constrained matrix factorization. BMC Bioinform. 2018, 19, 233. [Google Scholar] [CrossRef] [PubMed]
- Xuan, P.; Cao, Y.; Zhang, T.; Wang, X.; Pan, S.; Shen, T. Drug repositioning through integration of prior knowledge and projections of drugs and diseases. Bioinformatics 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, J.; O Suzek, T.; Zhang, J.; Wang, J.; Bryant, S.H. PubChem: A public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37, W623–W633. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, D.; Gong, Y.; Zhou, S.; Wang, J.; Zheng, N. Person re-identification by multi-channel parts-based cnn with improved triplet loss function. In Proceedings of the IEEE Conference on Computer Vision and Pattern recognition, Las Vegas, NV, USA, 26 June–1 July 2016; pp. 1335–1344. [Google Scholar]
- Nair, V.; Hinton, G.E. Rectified linear units improve restricted boltzmann machines. In Proceedings of the International Conference on International Conference on Machine Learning, Haifa, Israel, 21–24 June 2010. [Google Scholar]
- Elfwing, S.; Uchibe, E.; Doya, K. Sigmoid-weighted linear units for neural network function approximation in reinforcement learning. Neural Netw. 2018, 107, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Bahdanau, D.; Cho, K.; Bengio, Y. Neural machine translation by jointly learning to align and translate. In Proceedings of the International Conference on Learning Representations, Banff, AB, Canada, 14–16 April 2014. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Bell, R.M.; Koren, Y. Lessons from the Netflix prize challenge. ACM SIGKDD Explor. Newsl. 2007, 9, 75. [Google Scholar] [CrossRef]
- Hinton, G.E.; Srivastava, N.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R.R. Improving neural networks by preventing co-adaptation of feature detectors. In Proceedings of the International Conference on Learning Representations, Tsukuba, Japan, 11–15 November 2012. [Google Scholar]
- Xuan, P.; Sun, C.; Zhang, T.; Ye, Y.; Shen, T.; Dong, Y. Gradient Boosting Decision Tree-Based Method for Predicting Interactions Between Target Genes and Drugs. Front. Genet. 2019, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yang, S.; Zhang, X.; Li, J. Drug repositioning by integrating target information through a heterogeneous network model. Bioinformatics 2014, 30, 2923–2930. [Google Scholar] [CrossRef] [PubMed]
- Brock, H.; Moosbauer, W.; Gabriel, C.; Necek, S.; Bidal, D. Treatment of severe tetanus by continuous intrathecal infusion of baclofen. J. Neurol. Neurosurg. Psychiatry 1995, 59, 193–194. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | CGARDP | HGBI | AUC MBiRW | LRSSL | SCMFDD |
|---|---|---|---|---|---|
| ampicillin | 0.964 | 0.751 | 0.932 | 0.962 | 0.895 |
| cefepime | 0.990 | 0.910 | 0.970 | 0.971 | 0.914 |
| cefotaxime | 0.958 | 0.917 | 0.929 | 0.950 | 0.953 |
| cefotetan | 0.973 | 0.808 | 0.918 | 0.948 | 0.848 |
| cefoxitin | 0.880 | 0.890 | 0.912 | 0.979 | 0.894 |
| ceftazidime | 0.938 | 0.845 | 0.931 | 0.936 | 0.922 |
| ceftizoxime | 0.929 | 0.960 | 0.961 | 0.923 | 0.962 |
| ceftriaxone | 0.999 | 0.945 | 0.898 | 0.955 | 0.811 |
| ciprofloxacin | 0.905 | 0.811 | 0.813 | 0.928 | 0.820 |
| doxorubicin | 0.951 | 0.487 | 0.921 | 0.727 | 0.460 |
| erythromycin | 0.948 | 0.827 | 0.887 | 0.918 | 0.764 |
| itraconazole | 0.956 | 0.445 | 0.877 | 0.845 | 0.730 |
| levofloxacin | 0.898 | 0.943 | 0.975 | 0.964 | 0.872 |
| moxifloxacin | 0.992 | 0.812 | 0.948 | 0.957 | 0.932 |
| ofloxacin | 0.980 | 0.902 | 0.943 | 0.904 | 0.774 |
| Average AUC | 0.956 | 0.683 | 0.837 | 0.838 | 0.726 |
| Drug Name | CGARDP | HGBI | AUPR MBIRW | LRSSL | SCMFDD |
|---|---|---|---|---|---|
| ampicillin | 0.515 | 0.032 | 0.023 | 0.285 | 0.068 |
| cefepime | 0.766 | 0.163 | 0.315 | 0.625 | 0.054 |
| cefotaxime | 0.525 | 0.071 | 0.292 | 0.283 | 0.105 |
| cefotetan | 0.496 | 0.054 | 0.197 | 0.512 | 0.059 |
| cefoxitin | 0.420 | 0.151 | 0.394 | 0.286 | 0.065 |
| ceftazidime | 0.591 | 0.032 | 0.201 | 0.488 | 0.694 |
| ceftizoxime | 0.472 | 0.212 | 0.244 | 0.455 | 0.096 |
| ceftriaxone | 0.607 | 0.056 | 0.223 | 0.673 | 0.077 |
| ciprofloxacin | 0.429 | 0.082 | 0.118 | 0.280 | 0.064 |
| doxorubicin | 0.520 | 0.005 | 0.051 | 0.180 | 0.004 |
| erythromycin | 0.592 | 0.023 | 0.038 | 0.144 | 0.022 |
| itraconazole | 0.379 | 0.006 | 0.253 | 0.042 | 0.008 |
| levofloxacin | 0.212 | 0.136 | 0.071 | 0.539 | 0.098 |
| moxifloxacin | 0.735 | 0.049 | 0.650 | 0.384 | 0.088 |
| ofloxacin | 0.382 | 0.091 | 0.130 | 0.201 | 0.078 |
| Average AUC | 0.425 | 0.013 | 0.047 | 0.117 | 0.014 |
| p-Value between CGARDP and Another Method | HGBI | MBiRW | LRSSL | SCMFDD |
|---|---|---|---|---|
| p-value of ROC curve | 6.873 × 10−270 | 6.302 × 10−72 | 3.473 × 10−31 | 9.326 × 10−180 |
| p-value of PR curve | 4.365 × 10−40 | 7.332 × 10−30 | 2.321 × 10−12 | 3.265 × 10−60 |
| Drug Name | Rank | Disease Name | Description | Rank | Disease Name | Description |
|---|---|---|---|---|---|---|
| Ciprofloxacin | 1 | Conjunctivitis, Bacterial | Clinical Trials | 6 | Gram-Negative Bacterial Infections | Clinical Trials |
| 2 | Campylobacter Infections | CDC | 7 | Chlamydia Infections | Clinical Trials | |
| 3 | Anthrax | CTD, Clinical Trials | 8 | Pneumonia, Pneumocystis | PubChem | |
| 4 | Klebsiella Infections | CTD, Clinical Trials | 9 | Eye Infections, Bacterial | Clinical Trials | |
| 5 | Soft Tissue Infections | Clinical Trials | 10 | Acanthamoeba Keratitis | PubChem | |
| Ceftriaxone | 1 | Bone Diseases, Infectious | Clinical Trials | 6 | Tetanus | literature [38] |
| 2 | Panic Disorder | Drug Bank | 7 | Legionnaires Disease | Drug Bank | |
| 3 | Hepatitis B | Clinical Trials | 8 | Cytomegalovirus Infections | Drug Bank | |
| 4 | Respiratory Syncytial Virus Infections | PubChem | 9 | Respiration Disorders | Clinical Trials | |
| 5 | Maxillary Sinusitis | Drug Bank | 10 | Respiratory Distress Syndrome, Adult | Clinical Trials | |
| Ofloxacin | 1 | Corneal Ulcer | PubChem | 6 | Proteus Infections | CTD |
| 2 | Epididymitis | CDC | 7 | Urinary Bladder Neck Obstruction | Inferred candidate by 1 literature | |
| 3 | Otitis Externa | Drug Bank | 8 | Glaucoma, Angle-Closure | PubChem | |
| 4 | Tuberculosis, Pulmonary | CTD, clinical Trials | 9 | Urinary Bladder Diseases | Inferred candidate by 1 literature | |
| 5 | Urethral Diseases | PubChem | 10 | Trichomonas Vaginitis | clinical Trials | |
| Ampicillin | 1 | Burns | Inferred candidate by 3 literature | 6 | Candidiasis, Cutaneous | PubChem |
| 2 | Meningitis, Bacterial | CTD | 7 | Otitis Media, Suppurative | Drug Bank | |
| 3 | Pseudomonas Infections | CTD | 8 | Pneumonia, Bacterial | CTD, Clinical Trials | |
| 4 | Skin Diseases, Infectious | Clinical Trials | 9 | Proteus Infections | CTD | |
| 5 | Radiation Injuries, Experimental | Inferred candidate by 1 literature | 10 | Sarcoma, Ewings | Drug Bank | |
| Levofloxacin | 1 | Tuberculosis, Pulmonary | Clinical Trials | 6 | Listeriosis | Drug Bank |
| 2 | Histoplasmosis | Drug Bank | 7 | Soft Tissue Infections | CTD, Clinical Trials | |
| 3 | Pneumonia, Mycoplasma | Clinical Trials | 8 | Respiratory Tract Fistula | Drug Bank | |
| 4 | Bronchitis | Clinical Trials | 9 | Rhinitis | Drug Bank | |
| 5 | AIDS-Related Opportunistic Infections | Clinical Trials | 10 | Mouth Diseases | Clinical Trials |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xuan, P.; Zhao, L.; Zhang, T.; Ye, Y.; Zhang, Y. Inferring Drug-Related Diseases Based on Convolutional Neural Network and Gated Recurrent Unit. Molecules 2019, 24, 2712. https://doi.org/10.3390/molecules24152712
Xuan P, Zhao L, Zhang T, Ye Y, Zhang Y. Inferring Drug-Related Diseases Based on Convolutional Neural Network and Gated Recurrent Unit. Molecules. 2019; 24(15):2712. https://doi.org/10.3390/molecules24152712
Chicago/Turabian StyleXuan, Ping, Lianfeng Zhao, Tiangang Zhang, Yilin Ye, and Yan Zhang. 2019. "Inferring Drug-Related Diseases Based on Convolutional Neural Network and Gated Recurrent Unit" Molecules 24, no. 15: 2712. https://doi.org/10.3390/molecules24152712
APA StyleXuan, P., Zhao, L., Zhang, T., Ye, Y., & Zhang, Y. (2019). Inferring Drug-Related Diseases Based on Convolutional Neural Network and Gated Recurrent Unit. Molecules, 24(15), 2712. https://doi.org/10.3390/molecules24152712