
Cyclization of Single-Chain Fv Antibodies Markedly Suppressed Their Characteristic Aggregation Mediated by Inter-Chain VH-VL Interactions

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
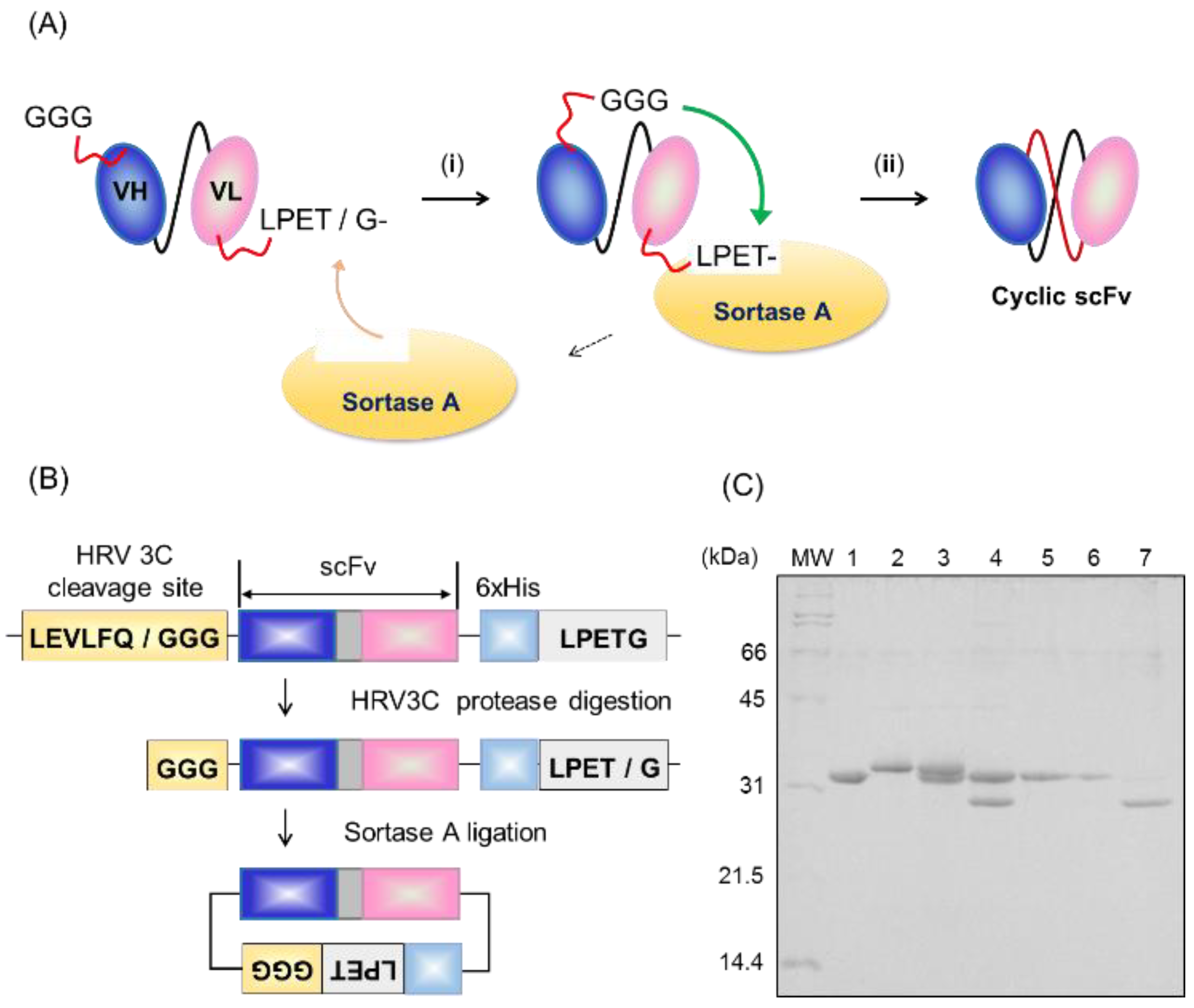
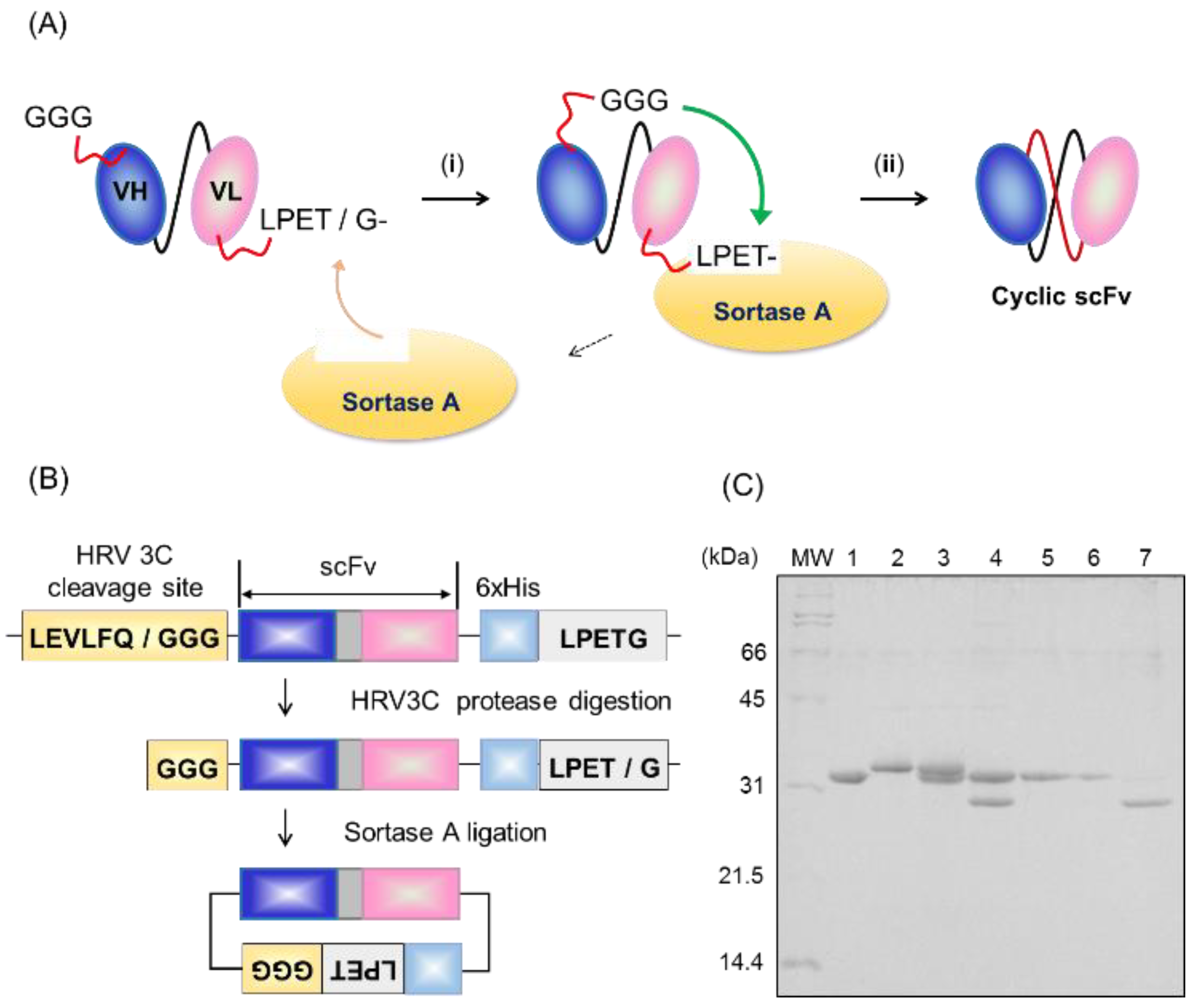
2.1. Preparation of Cyclic scFv by Sortase A Ligation
2.2. Evaluation of Open-Closed Dynamics by High-Speed Atomic Force Microscopy
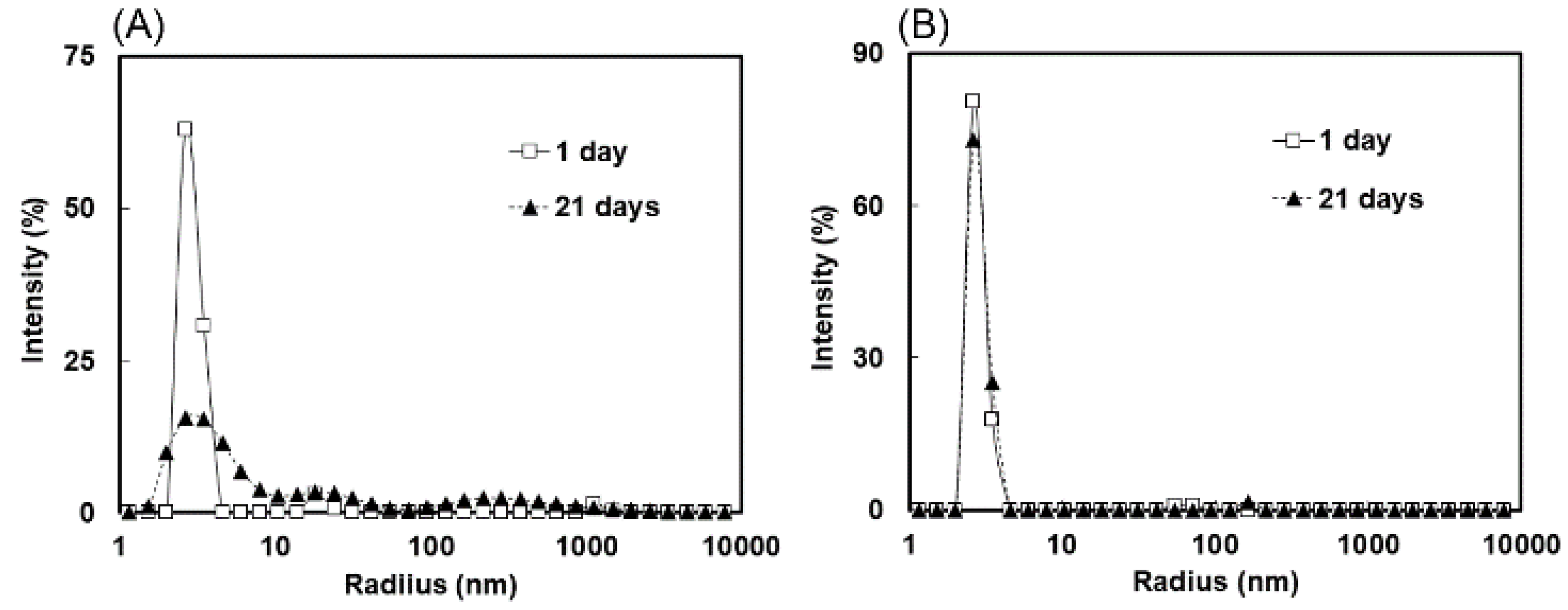
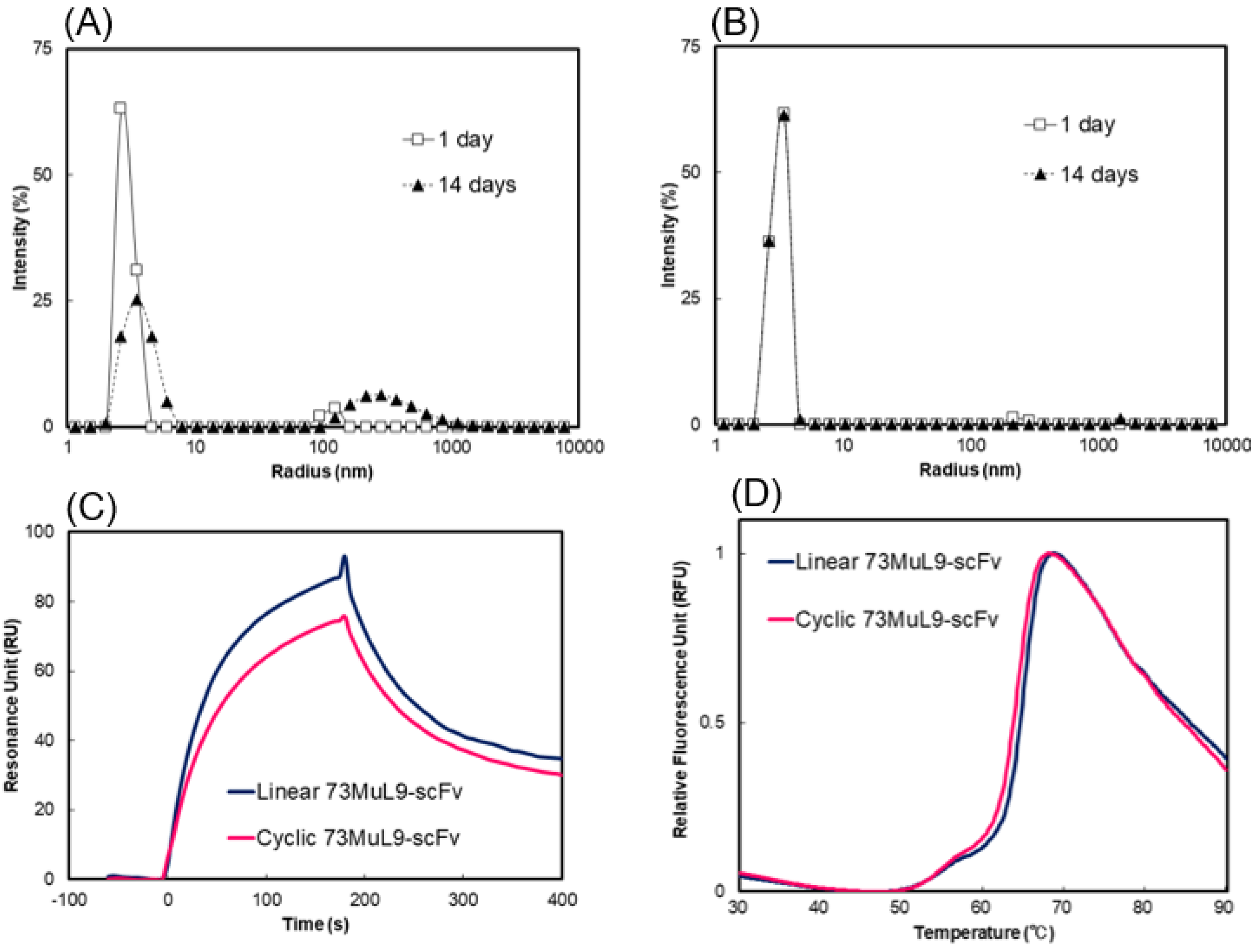
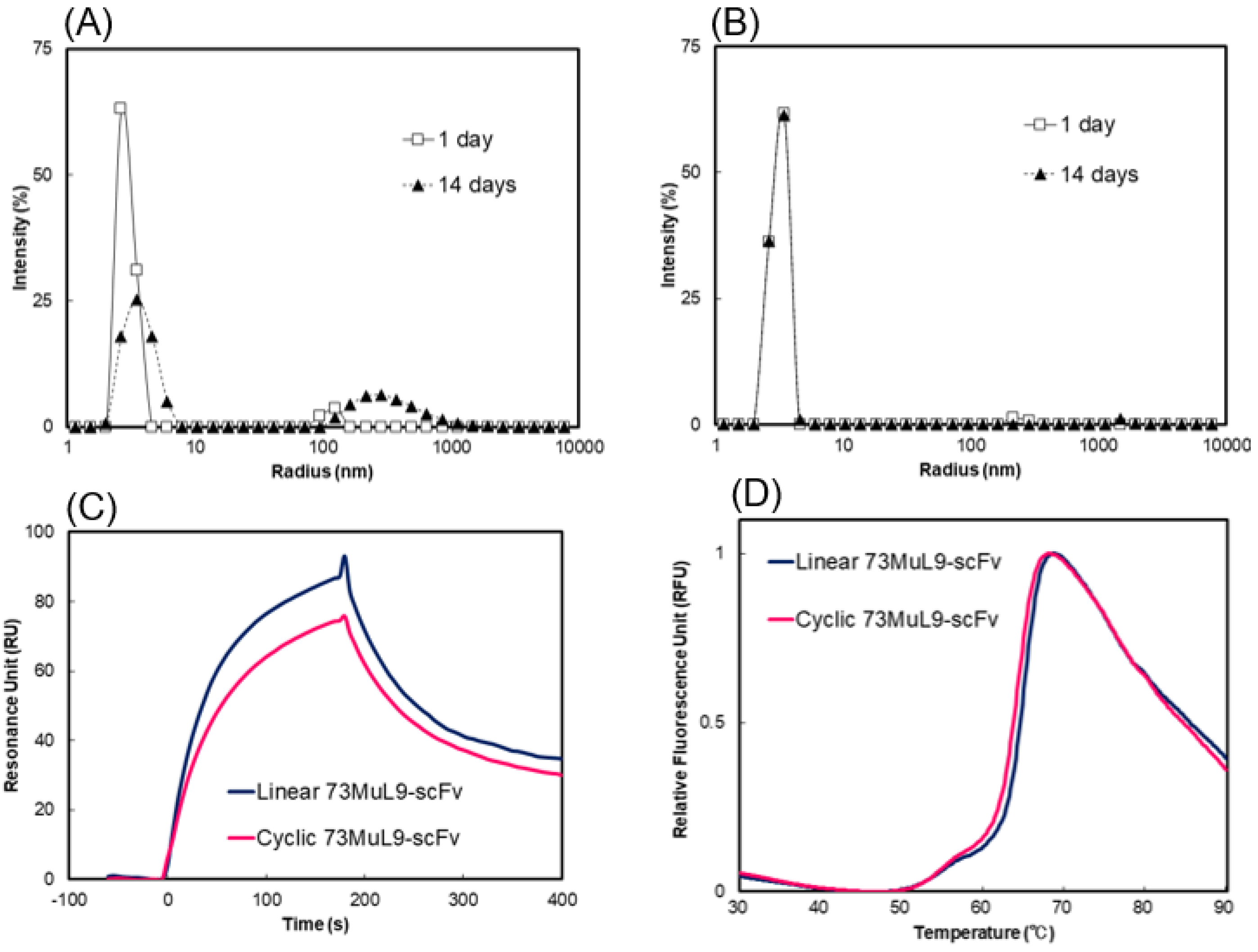
2.3. Measurement of Molecular Size Distributions by Dynamic Light Scattering
2.4. Evaluation of Affinity and Thermal Stability by Surface Plasmon Resonance and Differential Scanning Fluorometry
2.5. Generality of Cyclization for Suppression of Aggregation of scFv Antibodies
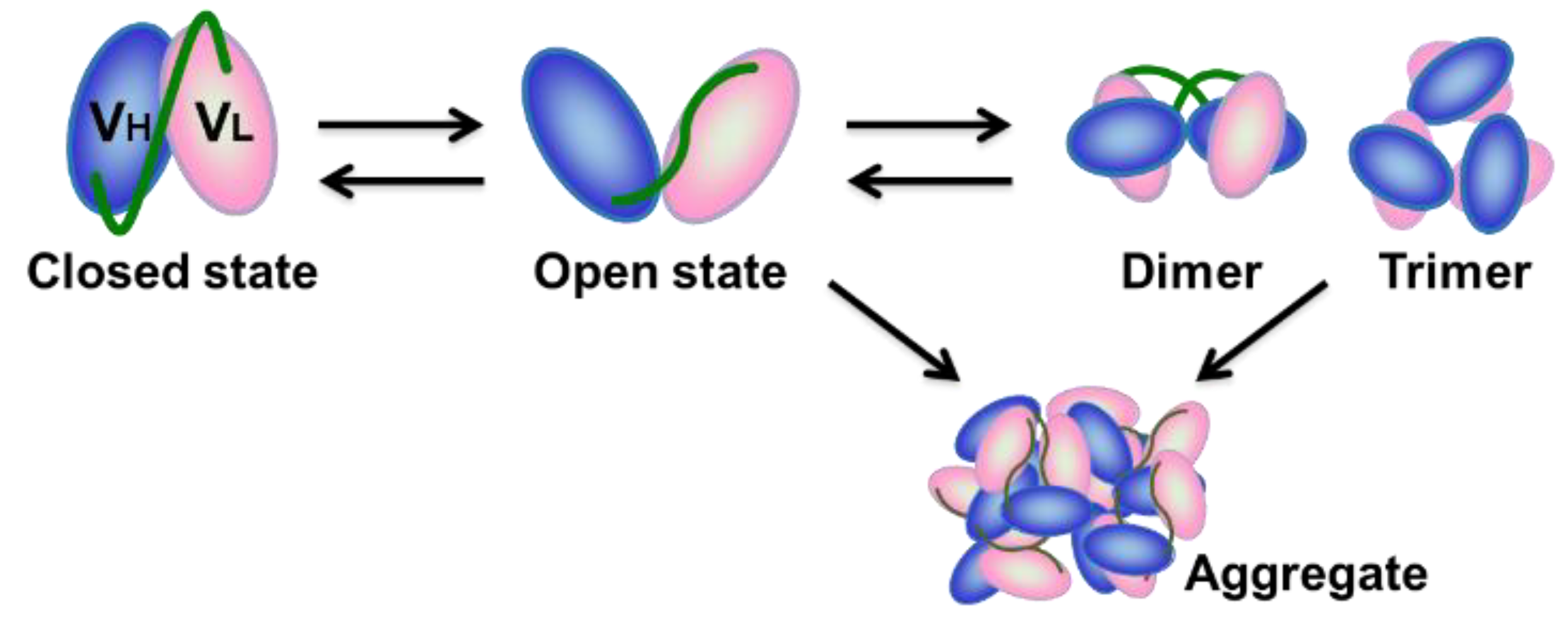
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Protein Ligation and Purification of the Product
4.3. Mass Spectrometry (LC-MS/MS)
4.4. Dynamic Light Scattering (DLS)
4.5. High-Speed Atomic Force Microscopy (HS-AFM)
4.6. Surface Plasmon Resonance (SPR)
4.7. Differential Scanning Fluorometry (DSF)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ducancel, F.; Muller, B.H. Molecular engineering of antibodies for therapeutic and diagnostic purposes. MAbs 2012, 4, 445–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larisa, J.; Geskin, M.D. Monoclonal antibodies. Dermatol. Clin. 2015, 33, 777–786. [Google Scholar]
- Gopinath, S.C.; Awazu, K.; Fujimaki, M.; Shimizu, K. Evaluation of anti-A/Udorn/307/1972 antibody specificity to influenza A/H3N2 viruses using an evanescent-field coupled waveguide-mode sensor. PLoS ONE 2013, 8, e81396. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, N.; Suwa, Y.; Uchida, M.; Kobashigawa, Y.; Yokoyama, H.; Morioka, H. Role of the mobility of antigen binding site in high affinity antibody elucidated by surface plasmon resonance. J. Biochem. 2017, 161, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Huston, J.S.; Levinson, D.; Mudgett-Hunter, M.; Tai, M.S.; Novotný, J.; Margolies, M.N.; Ridge, R.J.; Bruccoleri, R.E.; Haber, E.; Crea, R. Protein engineering of antibody binding sites: Recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc. Natl. Acad. Sci. USA 1988, 85, 5879–5883. [Google Scholar] [CrossRef] [PubMed]
- Jermutus, L.; Honegger, A.; Schwesinger, F.; Hanes, J.; Plückthun, A. Tailoring in vitro evolution for protein affinity or stability. Proc. Natl. Acad. Sci. USA 2000, 98, 75–80. [Google Scholar] [CrossRef]
- Plückthun, A.; Skerra, A. Expression of functional antibody Fv and Fab fragments in Escherichia coli. Methods Enzymol. 1989, 178, 497–515. [Google Scholar]
- Kobayashi, H.; Morioka, H.; Tobisawa, K.; Torizawa, T.; Kato, K.; Shimada, I.; Nikaido, O.; Stewart, J.D.; Ohtsuka, E. Probing the interaction between a high-affinity single-chain Fv and a pyrimidine (6-4) pyrimidone photodimer by site-directed mutagenesis. Biochemistry 1999, 38, 532–539. [Google Scholar] [CrossRef]
- Garber, K. Bispecific antibodies rise again. Nat. Rev. Drug Discov. 2014, 13, 799–801. [Google Scholar] [CrossRef]
- Kontermann, R.E.; Brinkmann, U. Bispecific antibodies. Drug Discov. Today 2015, 20, 838–847. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, A.; Jimeno, A. Bispecific antibodies for cancer therapy: A review. Pharmacol. Ther. 2018, 185, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Sedykh, S.E.; Prinz, V.V.; Buneva, V.N.; Nevinsky, G.A. Bispecific antibodies: Design, therapy, perspectives. Drug Des. Devel. Ther. 2018, 12, 195–208. [Google Scholar] [CrossRef]
- Arndt, K.M.; Müller, K.M.; Plückthun, A. Factors influencing the dimer to monomer transition of an antibody single-chain Fv fragment. Biochemistry 1998, 37, 12918–12926. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Boehm, M.K.; Chester, K.A.; Begent, R.H.; Perkins, S.J. Reversible dimer formation and stability of the anti-tumour single-chain Fv antibody MFE-23 by neutron scattering, analytical ultracentrifugation, and NMR and FT-IR spectroscopy. J. Mol. Biol. 2002, 320, 107–127. [Google Scholar] [CrossRef]
- Kortt, A.A.; Malby, R.L.; Caldwell, J.B.; Gruen, L.C.; Ivancic, N.; Lawrence, M.C.; Howlett, G.J.; Webster, R.G.; Hudson, P.J.; Colman, P.M. Recombinant anti-sialidase single-chain variable fragment antibody. Characterization, formation of dimer and higher-molecular-mass multimers and the solution of the crystal structure of the single-chain variable fragment/sialidase complex. Eur. J. Biochem. 1994, 221, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, N.; Noi, K.; Weng, L.; Kobashigawa, Y.; Miyazaki, H.; Wakeyama, Y.; Takaki, M.; Nakahara, Y.; Tatsuno, Y.; Uchida-Kamekura, M.; et al. Production of single-chain Fv antibodies specific for GA-Pyridine, an advanced glycation end-product (AGE), with reduced inter-domain motion. Molecules 2017, 22, 1695. [Google Scholar] [CrossRef] [PubMed]
- Bird, R.E.; Hardman, K.D.; Jacobson, J.W.; Johnson, S.; Kaufman, B.M.; Lee, S.M.; Lee, T.; Pope, S.H.; Riordan, G.S.; Whitlow, M. Single-chain antigen-binding proteins. Science 1988, 242, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Glockshuber, R.; Malia, M.; Pfitzinger, I.; Plückthun, A. A comparison of strategies to stabilize immunoglobulin Fv-fragments. Biochemistry 1990, 29, 1362–1367. [Google Scholar] [CrossRef]
- Weatherill, E.E.; Cain, K.L.; Heywood, S.P.; Compson, J.E.; Heads, J.T.; Adams, R.; Humphreys, D.P. Towards a universal disulphide stabilised single chain Fv format: Importance of interchain disulphide bond location and vL–vH orientation. Protein Eng. Des. Sel. 2012, 25, 321–329. [Google Scholar] [CrossRef]
- Young, N.M.; MacKenzie, C.R.; Narang, S.A.; Oomen, R.P.; Baenziger, J.E. Thermal stabilization of a single-chain Fv antibody fragment by introduction of a disulphide bond. FEBS Lett. 1995, 377, 135–139. [Google Scholar] [CrossRef] [Green Version]
- Wörn, A.; Plückthun, A. Different equilibrium stability behavior of ScFv fragments: Identification, classification, and improvement by protein engineering. Biochemistry 1999, 38, 8739–8750. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, V.; Pastan, I.; Kreitman, R.J. A form of anti-Tac (Fv) which is both single-chain and disulfide stabilized: Comparison with its single-chain and disulfide-stabilized homologs. Protein Eng. 1997, 10, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Dudgeon, K.; Rouet, R.; Kokmeijer, I.; Schofield, P.; Stolp, J.; Langley, D.; Stock, D.; Christ, D. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc. Natl. Acad. Sci. USA 2012, 109, 10879–10884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.; Honegger, A.; Plückthun, A. Selection for improved protein stability by phage display. J. Mol. Biol. 1999, 294, 163–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockmann, E.C.; Cooper, M.; Strömsten, N.; Vehniäinen, M.; Saviranta, P. Selecting for antibody scFv fragments with improved stability using phage display with denaturation under reducing conditions. J. Immunol. Methods 2005, 296, 159–170. [Google Scholar] [CrossRef]
- Suree, N.; Liew, C.K.; Villareal, V.A.; Thieu, W.; Fadeev, E.A.; Clemens, J.J.; Jung, M.E.; Clubb, R.T. The structure of the Staphylococcus aureus sortase-substrate complex reveals how the universally conserved LPXTG sorting signal is recognized. J. Biol. Chem. 2009, 284, 24465–24477. [Google Scholar] [CrossRef]
- Wu, Z.; Guo, Z. Sortase–Mediated Transpeptidation for Site–Specific Modification of Peptides, Glycopeptides, and Proteins. J. Carbohydr. Chem. 2012, 31, 48–66. [Google Scholar] [CrossRef]
- Ilangovan, U.; Ton-That, H.; Iwahara, J.; Schneewind, O.; Clubb, R.T. Structure of sortase, the transpeptidase that anchors proteins to the cell wall of Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2001, 98, 6056–6061. [Google Scholar] [CrossRef]
- Mao, H.; Hart, S.A.; Schink, A.; Pollok, B.A. Sortase-mediated protein ligation: A new method for protein engineering. J. Am. Chem. Soc. 2004, 126, 2670–2671. [Google Scholar] [CrossRef]
- Sato, Y.; Mukai, M.; Ueda, J.; Muraki, M.; Stasevich, T.J.; Horikoshi, N.; Kujirai, T.; Kita, H.; Kimura, T.; Hira, S.; et al. Genetically encoded system to track histone modification in vivo. Sci. Rep. 2013, 2436. [Google Scholar] [CrossRef]
- Torizawa, T.; Yamamoto, N.; Suzuki, T.; Nobuoka, K.; Komatsu, Y.; Morioka, H.; Nikaido, O.; Ohtsuka, E.; Kato, K.; Shimada, I. DNA binding mode of the Fab fragment of a monoclonal antibody specific for cyclobutane pyrimidine dimer. Nucleic Acids Res. 2000, 28, 944–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobashigawa, Y.; Kumeta, H.; Ogura, K.; Inagaki, F. Attachment of an NMR-invisible solubility enhancement tag using a sortase-mediated protein ligation method. J. Biomol. NMR 2009, 43, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Antos, J.M.; Popp, M.W.; Ernst, R.; Chew, G.L.; Spooner, E.; Ploegh, H.L. A straight path to circular proteins. J. Biol. Chem. 2009, 284, 16028–16036. [Google Scholar] [CrossRef] [PubMed]
- Popp, M.W.; Ploegh, H.L. Making and breaking peptide bonds: Protein engineering using sortase. Angew. Chem. Int. Ed. Engl. 2011, 50, 5024–5032. [Google Scholar] [CrossRef] [PubMed]
- Popp, M.W.; Dougan, S.K.; Chuang, T.Y.; Spooner, E.; Ploegh, H.L. Sortase-catalyzed transformations that improve the properties of cytokines. Proc. Natl. Acad. Sci. USA 2011, 108, 3169–3174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirakawa, H.; Ishikawa, S.; Nagamune, T. Ca2+-independent sortase-A exhibits high selective protein ligation activity in the cytoplasm of Escherichia coli. Biotechnol. J. 2015, 10, 1487–1492. [Google Scholar] [CrossRef] [PubMed]
- Ingenito, R.; Bianchi, E.; Fattori, D.; Pessi, A. Solid phase synthesis of peptide C-terminal thioesters by Fmoc/t-Bu chemistry. J. Org. Chem. 2004, 69, 4145–4151. [Google Scholar] [CrossRef]
- Tam, J.P.; Wong, C.T. Chemical synthesis of circular proteins. J. Biol. Chem. 2012, 287, 27020–27025. [Google Scholar] [CrossRef]
- Austin, J.; imura, R.H.; Woo, Y.H.; Camarero, J.A. In vivo biosynthesis of an Ala-scan library based on the cyclic peptide SFTI-1. Amino Acids 2010, 38, 1313–1322. [Google Scholar] [CrossRef]
- Scott, C.P.; Abel-Santos, E.; Wall, M.; Wahnon, D.C.; Benkovic, S.J. Production of cyclic peptides and proteins in vivo. Proc. Natl. Acad. Sci. USA 1999, 96, 13638–13643. [Google Scholar] [CrossRef]
- Nguyen, G.K.; Wang, S.; Qiu, Y.; Hemu, X.; Lian, Y.; Tam, J.P. Butelase 1 is an Asx-specific ligase enabling peptide macrocyclization and synthesis. Nat. Chem. Biol. 2014, 10, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Mikula, K.M.; Krumwiede, L.; Plückthun, A.; Iwai, H. Segmental isotopic labeling by asparaginyl endopeptidase-mediated protein ligation. J. Biomol. NMR 2018, 71, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.; Dorr, B.M.; Liu, D.R. A general strategy for the evolution of bond-forming enzymes using yeast display. Proc. Natl. Acad. Sci. USA 2011, 108, 11399–11404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, T.; Tomita, M.; Ishihama, Y. Phase transfer surfactant-aided trypsin digestion for membrane proteome analysis. J. Proteome Res. 2008, 7, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Saito, N.; Tomita, M.; Ishihama, Y. Unbiased quantitation of Escherichia coli membrane proteome using phase transfer surfactants. Mol. Cell Proteom. 2009, 8, 2770–2777. [Google Scholar] [CrossRef]
- Rappsilber, J.; Ishihama, Y.; Mann, M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003, 75, 663–670. [Google Scholar] [CrossRef]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef]
- Kobashigawa, Y.; Amano, S.; Yoza, K.; Himeno, R.; Amemiya, S.; Morioka, H.; Yokogawa, M.; Kumeta, H.; Schlessinger, J.; Inagaki, F. Nuclear magnetic resonance analysis of the conformational state of cancer mutant of fibroblast growth factor receptor 1 tyrosine kinase domain. Genes Cells 2016, 21, 350–357. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Not available. |



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamauchi, S.; Kobashigawa, Y.; Fukuda, N.; Teramoto, M.; Toyota, Y.; Liu, C.; Ikeguchi, Y.; Sato, T.; Sato, Y.; Kimura, H.; et al. Cyclization of Single-Chain Fv Antibodies Markedly Suppressed Their Characteristic Aggregation Mediated by Inter-Chain VH-VL Interactions. Molecules 2019, 24, 2620. https://doi.org/10.3390/molecules24142620
Yamauchi S, Kobashigawa Y, Fukuda N, Teramoto M, Toyota Y, Liu C, Ikeguchi Y, Sato T, Sato Y, Kimura H, et al. Cyclization of Single-Chain Fv Antibodies Markedly Suppressed Their Characteristic Aggregation Mediated by Inter-Chain VH-VL Interactions. Molecules. 2019; 24(14):2620. https://doi.org/10.3390/molecules24142620
Chicago/Turabian StyleYamauchi, Soichiro, Yoshihiro Kobashigawa, Natsuki Fukuda, Manaka Teramoto, Yuya Toyota, Chenjiang Liu, Yuka Ikeguchi, Takashi Sato, Yuko Sato, Hiroshi Kimura, and et al. 2019. "Cyclization of Single-Chain Fv Antibodies Markedly Suppressed Their Characteristic Aggregation Mediated by Inter-Chain VH-VL Interactions" Molecules 24, no. 14: 2620. https://doi.org/10.3390/molecules24142620