Design, Synthesis, and Biological Evaluation of EdAP, a 4′-Ethynyl-2′-Deoxyadenosine 5′-Monophosphate Analog, as a Potent Influenza a Inhibitor

Abstract
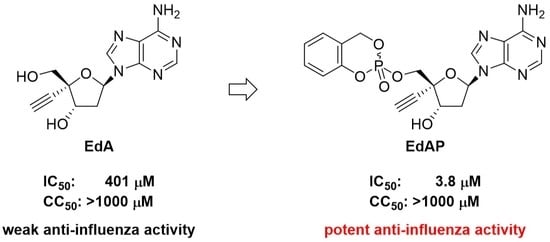
:
1. Introduction
2. Results and Discussion
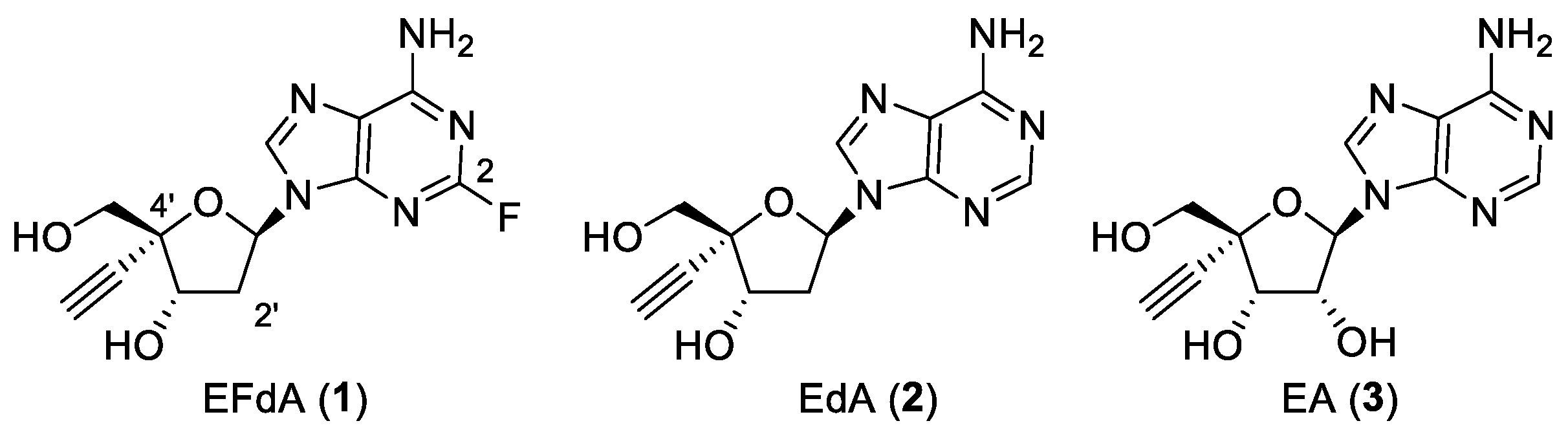
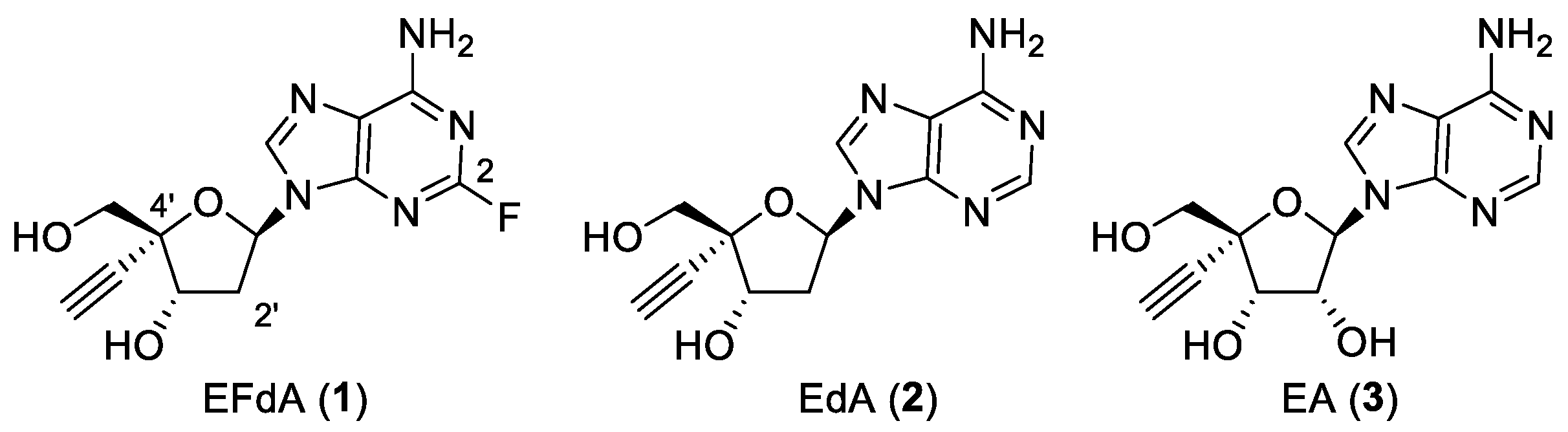
2.1. Effect of Compounds 1–3 on Anti-Influenza Activities
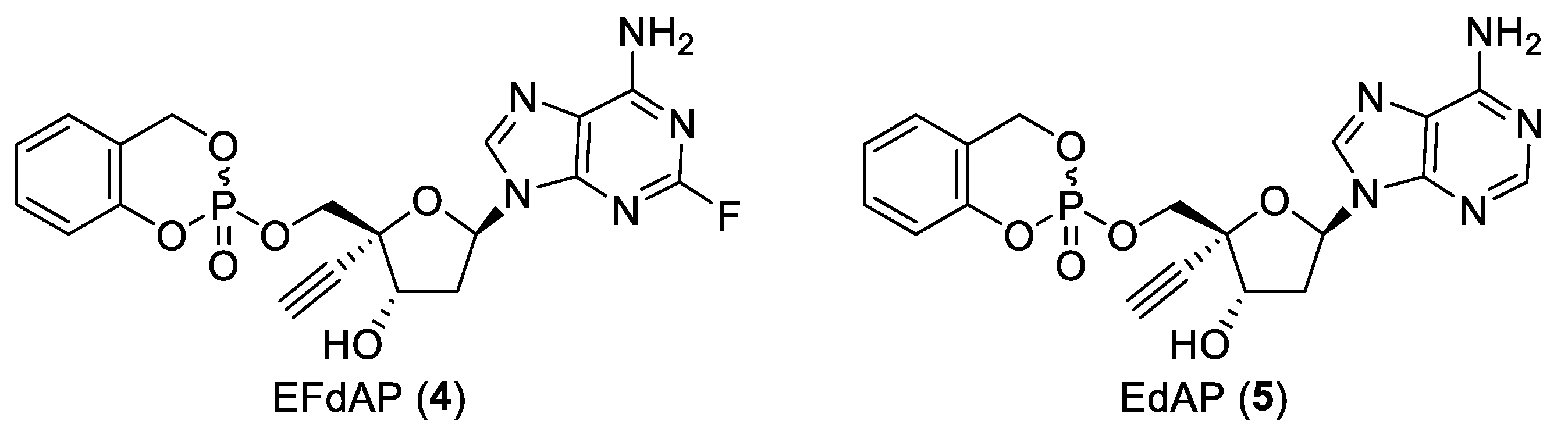
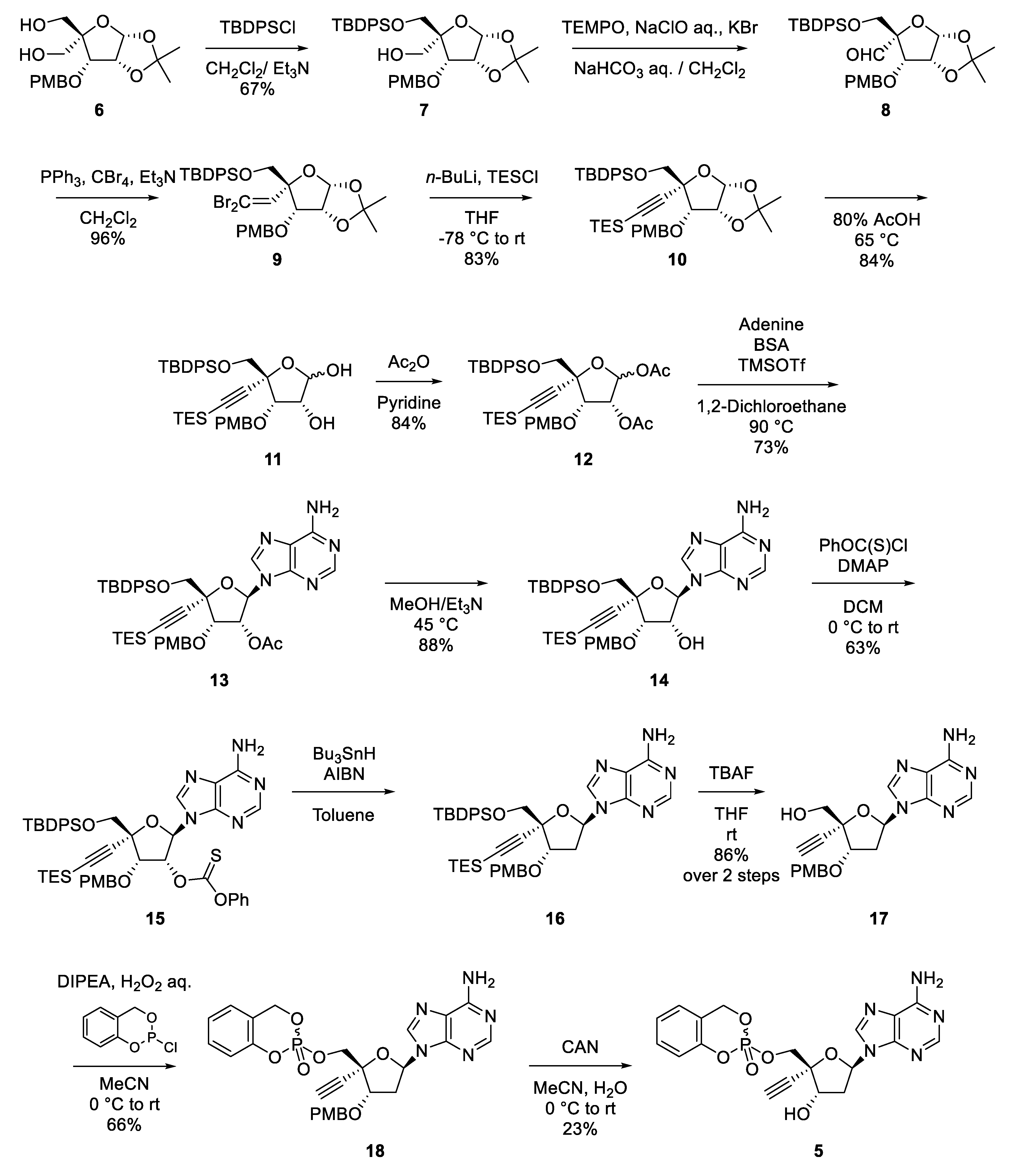
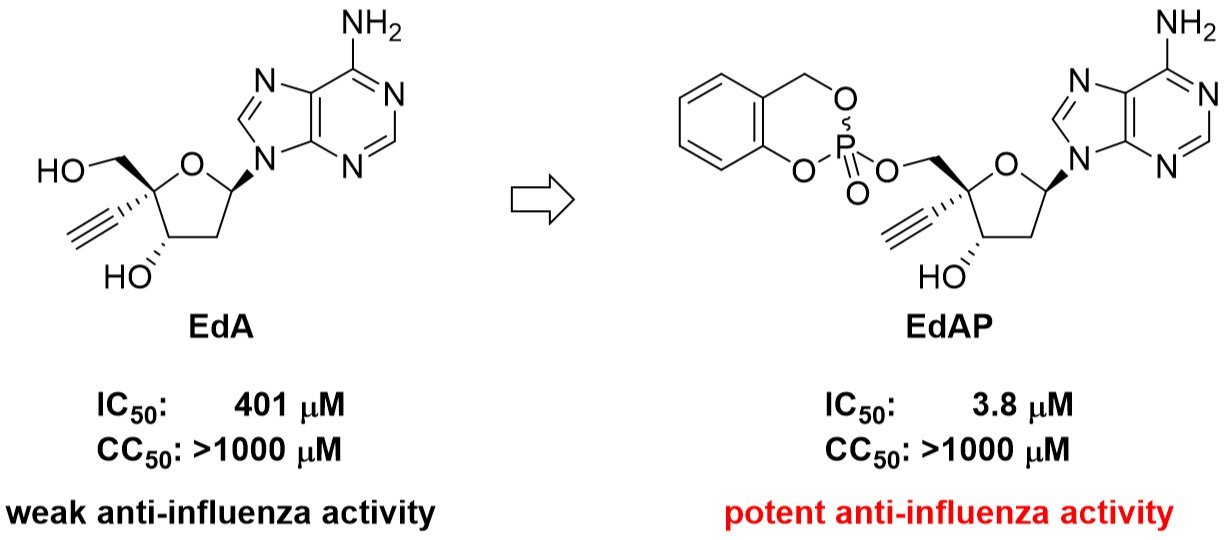
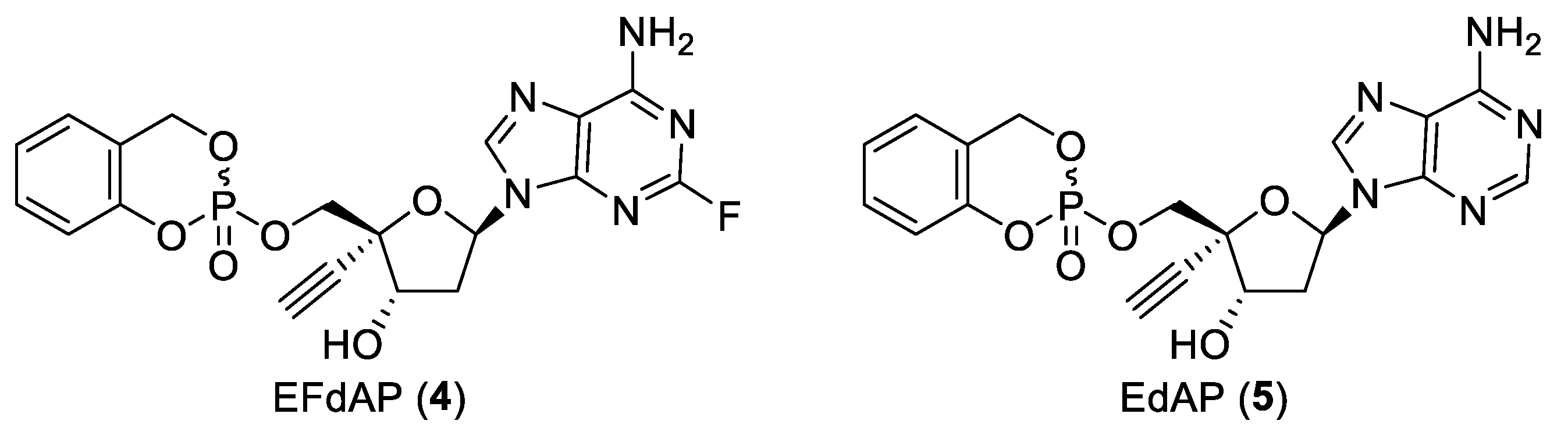
2.2. Synthesis of EdAP (5)
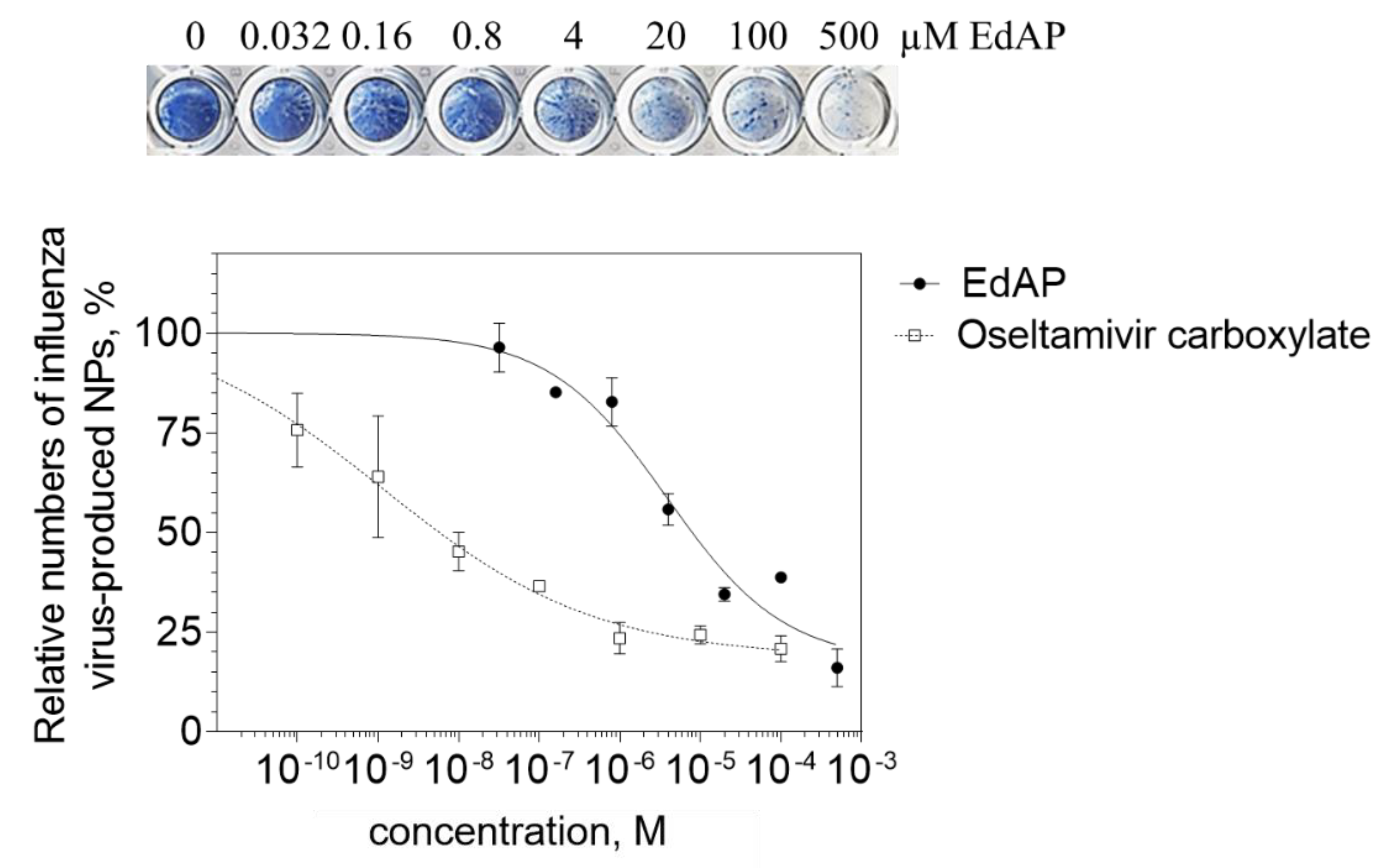
2.3. Anti-Influenza Activity of EdAP (5)
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis of ((3aR,5R,6S,6aR)-5-(((tert-butyldiphenylsilyl)oxy)methyl)-6-((4-methoxybenzyl)oxy)-2,2-dimethyltetrahydrofuro [2,3-d][1,3]dioxol-5-yl)methanol (7)
3.1.2. Synthesis of tert-butyl(((3aR,5R,6S,6aR)-5-(2,2-dibromovinyl)-6-((4-methoxybenzyl)oxy)-2,2- dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methoxy)diphenylsilane (9)
3.1.3. Synthesis of tert-butyl(((3aR,5R,6S,6aR)-6-((4-methoxybenzyl)oxy)-2,2-dimethyl-5-((triethylsilyl) ethynyl)tetrahydrofuro[2,3-d][1,3]dioxol-5-yl)methoxy)diphenylsilane (10)
3.1.4. Synthesis of (3R,4S,5R)-5-(((tert-butyldiphenylsilyl)oxy)methyl)-4-((4-methoxybenzyl)oxy)-5- ((triethylsilyl)ethynyl)tetrahydrofuran-2,3-diyl diacetate (12)
3.1.5. Synthesis of (2R,3R,4S,5R)-2-(6-amino-9H-purin-9-yl)-5-(((tert-butyldiphenylsilyl)oxy)methyl)-4- ((4-methoxybenzyl)oxy)-5-((triethylsilyl)ethynyl)tetrahydrofuran-3-yl acetate (13)
3.1.6. Synthesis of (2R,3R,4S,5R)-2-(6-amino-9H-purin-9-yl)-5-(((tert-butyldiphenylsilyl)oxy)methyl)-4-((4- methoxybenzyl)oxy)-5-((triethylsilyl)ethynyl)tetrahydrofuran-3-ol (14)
3.1.7. Synthesis of O-((2R,3R,4S,5R)-2-(6-amino-9H-purin-9-yl)-5-(((tert-butyldiphenylsilyl)oxy)methyl)-4- ((4-methoxybenzyl)oxy)-5-((triethylsilyl)ethynyl)tetrahydrofuran-3-yl) O-phenyl carbonothioate (15)
3.1.8. Synthesis of 9-((2R,4S,5R)-5-(((tert-butyldiphenylsilyl)oxy)methyl)-4-((4-methoxybenzyl)oxy)-5-((triethylsilyl)ethynyl)tetrahydrofuran-2-yl)-9H-purin-6-amine (17)
3.1.9. Synthesis of 2-(((2R,3S,5R)-5-(6-amino-9H-purin-9-yl)-2-ethynyl-3-((4-methoxybenzyl)oxy) tetrahydrofuran-2-yl)methoxy)-4H-benzo[d][1,3,2]dioxaphosphinine 2-oxide (18)
3.1.10. Synthesis of 2-(((2R,3S,5R)-5-(6-amino-9H-purin-9-yl)-2-ethynyl-3-hydroxytetrahydrofuran-2-yl) methoxy)-4H-benzo[d][1,3,2]dioxaphosphinine 2-oxide (5)
3.2. Biological Assays
3.2.1. Cells
3.2.2. Virus
3.2.3. Cytotoxicity Assay
3.2.4. Influenza Growth Inhibition Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Global Influenza Strategy 2019–2030; World Health Organization: Geneva, Switzerland, 2019.
- Stevaert, A.; Naesens, L. The influenza virus polymerase complex: An update on its structure, functions, and significance for antiviral drug design. Med. Res. Rev. 2016, 36, 1127–1173. [Google Scholar] [CrossRef] [PubMed]
- Laborda, P.; Wang, S.-Y.; Voglmeir, J. Influenza Neuraminidase Inhibitors: Synthetic Approaches, Derivatives and Biological Activity. Molecules 2016, 21, 1513. [Google Scholar] [CrossRef] [PubMed]
- Das, K. Antivirals Targeting Influenza A Virus. J. Med. Chem. 2012, 14, 6263–6277. [Google Scholar] [CrossRef] [PubMed]
- Kohgo, S.; Ohrui, H.; Kodama, E.; Matsuoka, M.; Mitsuya, H. 4′-c-Substituted-2-Haloadenosine Derivative. U.S. Patent 2502199, 4 March 2005. [Google Scholar]
- Kohgo, S.; Yamada, K.; Kitano, K.; Iwai, Y.; Sakata, S.; Ashida, N.; Hayakawa, H.; Nameki, D.; Kodama, E.; Matsupka, M.; et al. Design, efficient synthesis, and anti-HIV activity of 4′-C-cyano- and 4′-C-ethynyl-2′-deoxy purine nucleosides. Nucleosides Nucleotides Nucleic Acids 2004, 23, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Ohrui, H. 2′-deoxy-4′-C-ethynyl-2-fluoroadenosine, a nucleoside reverse transcriptase inhibitor, is highly potent against all human immunodeficiency viruses type 1 and has low toxicity. Chem. Rec. 2006, 6, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Ohrui, H.; Hayakawa, H.; Kohgo, S.; Matsuoka, M.; Kodama, E.; Mitsuya, H. Creation of Low Toxic Reverse-transcriptase Inhibitory Nucleosides that Prevent the Emergence of Drug-resistant HIV Variants. J. Synth. Org. Chem. Jpn. 2006, 64, 716–723. [Google Scholar] [CrossRef]
- Ohrui, H.; Kohgo, S.; Hayakawa, H.; Kodama, E.; Matsuoka, M.; Nakata, T.; Mitsuya, H. 2′-deoxy-4′-C-ethynyl-2-fluoroadenosine: A nucleoside reverse transcriptase inhibitor with highly potent activity against wide spectrum of HIV-1 strains, favorable toxic profiles, and stability in plasma. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1543–1546. [Google Scholar] [CrossRef] [PubMed]
- Michailidis, E.; Marchand, B.; Kodama, E.; Singh, K.; Matsuoka, M.; Kirby, K.A.; Ryan, E.M.; Sawani, A.M.; Nagy, E.; Ashida, N.; et al. Mechanism of inhibition of HIV-1 reverse transcriptase by 4′-Ethynyl-2-fluoro-2′-deoxyadenosine triphosphate, a translocation-defective reverse transcriptase inhibitor. J. Biol. Chem. 2009, 284, 35681–35691. [Google Scholar] [CrossRef] [PubMed]
- Ohrui, H. Development of modified nucleosides that have supremely high anti-HIV activity and low toxicity and prevent the emergence of resistant HIV mutants. Proc. Jpn. Acad. Ser. B 2011, 87, 53–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michailidis, E.; Huber, A.D.; Ryan, E.M.; Ong, Y.T.; Leslie, M.D.; Matzek, K.B.; Singh, K.; Marchand, B.; Hagedorn, A.N.; Kirby, K.A.; et al. 4′-Ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) inhibits HIV-1 reverse transcriptase with multiple mechanisms. J. Biol. Chem. 2014, 289, 24533–24548. [Google Scholar] [CrossRef] [PubMed]
- Kodama, E.; Kohgo, S.; Kitano, K.; Machida, H.; Gatanaga, H.; Shigeta, S.; Matsuoka, M.; Ohrui, H.; Mitsuya, H. 4′-Ethynyl nucleoside analogs: Potent inhibitors of multidrug-resistant human immunodeficiency virus variants in vitro. Antimicrob. Agents Chemother. 2001, 45, 1539–1546. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, A.; Kodama, E.; Sarafianos, S.G.; Sakagami, Y.; Kohgo, S.; Kitano, K.; Ashida, N.; Iwai, Y.; Hayakawa, H.; Nakata, H.; et al. 2008. 2′-Deoxy-4′-C-ethynyl-2-halo-adenosines active against drug-resistant human immunodeficiency virus type 1 variants. Int. J. Biochem. Cell Biol. 2008, 40, 2410–2420. [Google Scholar] [CrossRef] [PubMed]
- Michailidis, E.; Ryan, E.M.; Hachiya, A.; Kirby, K.A.; Marchand, B.; Leslie, M.D.; Huber, A.D.; Ong, Y.T.; Jackson, J.C.; Singh, K.; et al. Hypersusceptibility mechanism of Tenofovir-resistant HIV to EFdA. Retrovirology 2013, 10, 65. [Google Scholar] [CrossRef] [PubMed]
- Maeada, K.; Desai, D.V.; Aoki, M.; Nakata, H.; Kodama, E.N.; Mitsuya, H. Delayed emergence of HIV-1 variants resistant to 4′-ethynyl-2-fluoro-2′-deoxyadenosine: Comparative sequential passage study with lamivudine, tenofovir, emtricitabine and BMS-986001. Antivir. Ther. 2014, 19, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Wu, V.H.; Smith, R.A.; Masoum, S.; Raugi, D.N.; Ba, S.; Seydi, M.; Grobler, J.A.; Gottlieb, G.S. MK-8591 (4′-ethynyl-2-fluoro-2′-deoxyadenosine) exhibits potent activity against HIV-2 isolates and drug-resistant HIV-2 mutants in culture. Antimicrob. Agents Chemother. 2017, 61, e00744-17. [Google Scholar] [CrossRef]
- Njenda, D.T.; Aralaguppe, S.G.; Singh, K.; Rao, R.; Sönnerborg, A.; Sarafianos, S.G.; Neogi, U. Antiretroviral potency of 4′-ethnyl-2′-fluoro-2′-deoxyadenosine, tenofovir alafenamide and second-generation NNRTIs across diverse HIV-1 subtypes. J. Antimicrob. Chemother. 2018, 73, 2721–2728. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, Y.; Das, D.; Kohgo, S.; Hayashi, H.; Delino, N.S.; Sarafianos, S.G.; Mitsuya, H.; Maeda, K. The High Genetic Barrier of EFdA/MK-8591 Stems from Strong Interactions with the Active Site of Drug-Resistant HIV-1 Reverse Transcriptase. Cell Chem. Biol. 2018, 25, 1268–1278. [Google Scholar]
- Murphey-Corb, M.; Rajakumar, P.; Michael, H.; Nyaundi, J.; Didier, P.J.; Reeve, A.B.; Mitsuya, H.; Sarafianos, S.G.; Parniak, M.A. Response of simian immunodeficiency virus to the novel nucleoside reverse transcriptase inhibitor 4′-ethynyl-2-fluoro-2′-deoxyadenosine in vitro and in vivo. Antimicrob. Agents Chemother. 2012, 56, 4707–4712. [Google Scholar] [CrossRef]
- Hachiya, A.; Reeve, A.B.; Marchand, B.; Michailidis, E.; Ong, Y.T.; Kirby, K.A.; Leslie, M.D.; Oka, S.; Kodama, E.N.; Rohan, L.C.; et al. Evaluation of combinations of 4′-ethynyl-2-fluoro-2′-deoxyadenosine with clinically used antiretroviral drugs. Antimicrob. Agents Chemother. 2013, 57, 4554–4558. [Google Scholar] [CrossRef]
- Zhang, W.; Parniak, M.A.; Mitsuya, H.; Sarafianos, S.G.; Graebing, P.W.; Rohan, L.C. Preformulation studies of EFdA, a novel nucleoside reverse transcriptase inhibitor for HIV prevention. Drug Dev. Ind. Pharm. 2014, 40, 1101–1111. [Google Scholar] [CrossRef]
- Stoddart, C.A.; Galkina, S.A.; Joshi, P.; Kosikova, G.; Moreno, M.E.; Rivera, J.M.; Sloan, B.; Reeve, A.B.; Sarafianos, S.G.; Murphey-Corb, M.; et al. Oral administration of the nucleoside EFdA (4′-ethynyl-2-fluoro-2′-deoxyadenosine) provides rapid suppression of HIV viremia in humanized mice and favorable pharmacokinetic properties in mice and the rhesus macaque. Antimicrob. Agents Chemother. 2015, 59, 4190–4198. [Google Scholar] [CrossRef]
- Markowitz, M.; Sarafianos, S.G. 4′-Ethynyl-2-fluoro-2′-deoxyadenosine, MK-8591: A novel HIV-1 reverse transcriptase translocation inhibitor. Curr. Opin. HIV AIDS 2018, 13, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Ohrui, H.; Kohgo, S.; Kitano, K.; Sakata, S.; Kodama, E.; Yoshimura, K.; Matsuoka, M.; Shigeta, S.; Mitsuya, H. Syntheses of 4′-C-ethynyl-beta-D-arabino- and 4′-C-ethynyl-2′-deoxy-beta-D-ribo- pentofuranosylpyrimidines and -purines and evaluation of their anti-HIV activity. J. Med. Chem. 2000, 43, 4516–4525. [Google Scholar] [CrossRef] [PubMed]
- Sriwilaijaroen, N.; Wilairat, P.; Hiramatsu, H.; Takahashi, T.; Suzuki, T.; Ito, M.; Ito, Y.; Tashiro, M.; Suzuki, Y. Mechanisms of the action of povidone-iodine against human and avian influenza A viruses: Its effects on hemagglutination and sialidase activities. Virol. J. 2009, 6, 124. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, F.; Takeuchi, T.; Kamisuki, S.; Ohrui, H.; Suzuki, Y. Japanese Kokai Tokkyo Koho. JP Patent 2015,151,346 A, 12 February 2015. [Google Scholar]
- Meier, C. 2-Nucleos-5′-O-yl-4H-1,3,2-benzodioxaphos-phinin-2-oxides—A new concept for lipophilic, potential prodrugs of biologically active nucleoside monophosphates. Angew. Chem. Int. Ed. Engl. 1996, 35, 70–72. [Google Scholar] [CrossRef]
- Meier, C.; Knispel, T.; Clercq, E.D.; Balzarini, J. cycloSal-Pronucleotides of 2′,3′-dideoxyadenosine and 2′, 3′-dideoxy-2′,3′-didehydroadenosine: Synthesis and antiviral evaluation of a highly efficient nucleotide delivery system. J. Med. Chem. 1999, 42, 1604–1614. [Google Scholar] [CrossRef] [PubMed]
- Meier, C.; Balzarini, J. Application of the cycloSal-prodrug approach for improving the biological potential of phosphorylated biomolecules. Antoviral Res. 2006, 71, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, R.; Imanishi, T.; Kohgo, S.; Horie, H.; Ohrui, H. Synthesis of 4′-C-ethynyl-β-D-ribo-pentofuranosyl pyrimidines. Biosci. Biotechnol. Biochem. 1999, 63, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J.; Fuchs, P.L. A synthetic method for formyl→ethynyl conversion (RCHO→RC≡CH or RC≡CR′). Tetrahedron Lett. 1972, 36, 3769–3772. [Google Scholar] [CrossRef]
- Leonard, N.J.; Laursen, R.A. The synthesis of 3-β-d-ribofuranosyladenine. J. Am. Chem. Soc. 1963, 85, 2026–2028. [Google Scholar] [CrossRef]
- Leonard, N.J.; Laursen, R.A. Synthesis of 3-β-d-ribofuranosyladenine and (3-β-d-ribofuranosyladenine)-5′-phosphate. Biochemistry 1965, 4, 354–365. [Google Scholar] [CrossRef]
- Barton, D.H.R.; McCombie, S.W. A new method for the deoxygenation of secondary alcohols. J. Chem. Soc. Perkin. Trans. 1975, 1, 1574–1585. [Google Scholar] [CrossRef]
- Crich, D.; Quintero, L. Radical chemistry associated with the thiocarbonyl group. Chem. Rev. 1989, 89, 1413–1432. [Google Scholar] [CrossRef]
- Hayakawa, H.; Kohgo, S.; Kitano, K.; Ashida, N.; Kodama, E.; Mitsuya, H.; Ohrui, H. Potential of 4′-C-substituted nucleosides for the treatment of HIV-1. Antivir. Chem. Chemother. 2004, 15, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Sriwilaijaroen, N.; Kadowaki, A.; Onishi, Y.; Gato, N.; Ujike, M.; Odagiri, T.; Tashiro, M.; Suzuki, Y. Mumefural and related HMF derivatives from Japanese apricot fruit juice concentrate show multiple inhibitory effects on pandemic influenza A (H1N1) virus. Food Chem. 2011, 127, 1–9. [Google Scholar] [CrossRef]
- Sriwilaijaroen, N.; Magesh, S.; Imamura, A.; Ando, H.; Ishida, H.; Sakai, M.; Ishitsubo, E.; Hori, T.; Moriya, S.; Ishikawa, T.; et al. A novel potent and highly specific inhibitor against influenza viral N1-N9 neuraminidases: Insight into neuraminidase-inhibitor interactions. J. Med. Chem. 2016, 59, 4563–4577. [Google Scholar] [CrossRef] [PubMed]
- Sriwilaijaroen, N.; Suzuki, K.; Takashita, E.; Hiramatsu, H.; Kanie, O.; Suzuki, Y. 6SLN-lipo PGA specifically catches (coats) human influenza virus and synergizes neuraminidase-targeting drugs for human influenza therapeutic potential. J. Antimicrob. Chemother. 2015, 70, 2797–2809. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H1N1 Influenza A Virus | MDCK–SIAT1 | |
|---|---|---|
| Compound | IC50 (μM) | CC50 1 (μM) |
| 1 | 63 | >1000 |
| 2 | 401 | >1000 |
| 3 | >1000 | >1000 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeuchi, T.; Sriwilaijaroen, N.; Sakuraba, A.; Hayashi, E.; Kamisuki, S.; Suzuki, Y.; Ohrui, H.; Sugawara, F. Design, Synthesis, and Biological Evaluation of EdAP, a 4′-Ethynyl-2′-Deoxyadenosine 5′-Monophosphate Analog, as a Potent Influenza a Inhibitor. Molecules 2019, 24, 2603. https://doi.org/10.3390/molecules24142603
Takeuchi T, Sriwilaijaroen N, Sakuraba A, Hayashi E, Kamisuki S, Suzuki Y, Ohrui H, Sugawara F. Design, Synthesis, and Biological Evaluation of EdAP, a 4′-Ethynyl-2′-Deoxyadenosine 5′-Monophosphate Analog, as a Potent Influenza a Inhibitor. Molecules. 2019; 24(14):2603. https://doi.org/10.3390/molecules24142603
Chicago/Turabian StyleTakeuchi, Toshifumi, Nongluk Sriwilaijaroen, Ayako Sakuraba, Ei Hayashi, Shinji Kamisuki, Yasuo Suzuki, Hiroshi Ohrui, and Fumio Sugawara. 2019. "Design, Synthesis, and Biological Evaluation of EdAP, a 4′-Ethynyl-2′-Deoxyadenosine 5′-Monophosphate Analog, as a Potent Influenza a Inhibitor" Molecules 24, no. 14: 2603. https://doi.org/10.3390/molecules24142603