Development and Validation of a Method for the Analysis of Bisoprolol and Atenolol in Human Bone

 ,
,
Abstract
:1. Introduction
2. Results
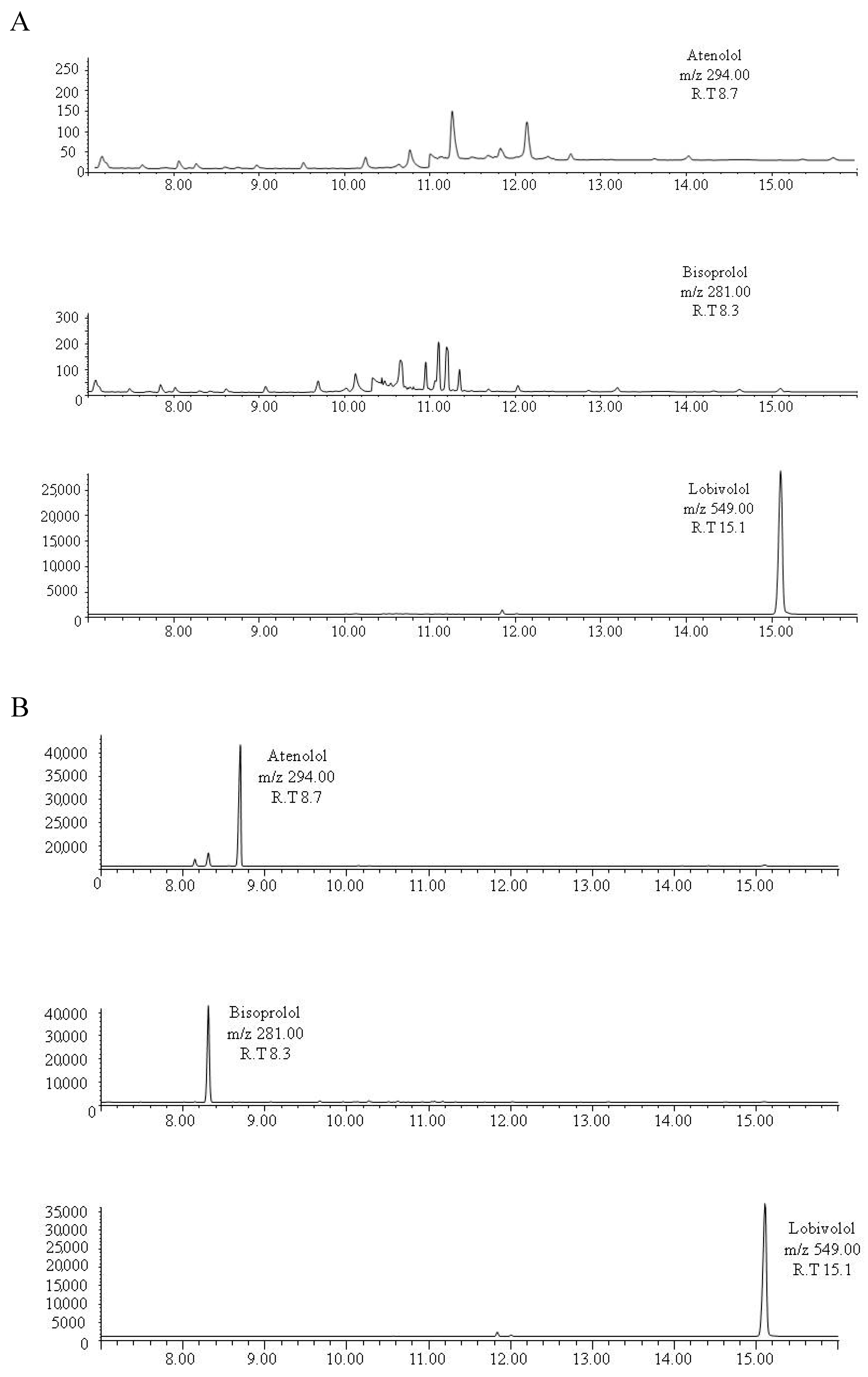
2.1. Gas Chromatography–Mass Spectrometry
2.2. Validation Results
2.3. Application to Real Samples
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Bone Samples
4.3. Preparation of Standard Solutions
4.4. Sample Preparation
4.5. Solid-Phase Extraction
4.6. Gas Chromatography–Mass Spectrometry Analysis
4.7. Method Validation
4.8. Expression of Analyte Levels
Author Contributions
Funding
Conflicts of Interest
References
- McGrath, K.K.; Jenkins, A.J. Detection of drugs of forensic importance in post-mortem bone. Am. J. Forensic Med. Pathol. 2009, 30, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Desrosiers, N.A.; Watterson, J.H.; Dean, D.E.; Wyman, J.F. Detection of amitriptyline and citalopram and metabolites in porcine bones following extended outdoor decomposition. J. Forensic Sci. 2012, 57, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Watterson, J. Challenges in forensic toxicology of skeletonised human remains. Analyst 2006, 131, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Guillot, E.; de Mazancourt, P.; Durigon, M.; Alvarez, J.C. Morphine and 6-acetylmorphine concentrations in blood, brain, spinal cord, bone marrow and bone after lethal acute or chronic diacetylmorphine administration to mice. Forensic Sci. Int. 2007, 166, 139–144. [Google Scholar] [CrossRef]
- Raikos, N.; Tsoukali, H.; Njau, S.N. Determination of opiates in postmortem bone and bone marrow. Forensic Sci. Int. 2009, 123, 140–141. [Google Scholar] [CrossRef]
- Watterson, J.H.; Donohue, J.P.; Betit, C.C. Comparison of relative distribution of ketamine and norketamine in decomposed skeletal tissues following single and repeated exposures. J. Anal. Toxicol. 2012, 36, 429–433. [Google Scholar] [CrossRef]
- Watterson, J.H.; Cornthwaite, H.M. Discrimination between patterns of drug exposure by toxicological analysis of decomposed skeletal tissues, Part II: Amitriptyline and Citalopram. J. Anal. Toxicol. 2013, 37, 565–572. [Google Scholar] [CrossRef]
- Watterson, J.H.; Desrosiers, N.A.; Betit, C.C.; Dean, D.; Wyman, J.F. Relative distribution of drugs in decomposed skeletal tissues. J. Anal. Toxicol. 2010, 34, 510–515. [Google Scholar] [CrossRef]
- Watterson, J.H.; Donohue, J.P. Relative distribution of ketamine and norketamine in skeletal tissues following various periods of decomposition. J. Anal. Toxicol. 2011, 35, 52–458. [Google Scholar] [CrossRef]
- Watterson, J.H.; Desrosiers, N.A. Microwave-assisted extraction in the study of the effect of dose-death interval on meperidine detection in skeletal tissues. Forensic Sci. Int. 2011, 207, 40–45. [Google Scholar] [CrossRef]
- Desrosiers, N.A.; Betit, C.C.; Watterson, J.H. Microwave-assisted extraction in toxicological screening of skeletal tissues. Forensic Sci. Int. 2009, 188, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Cornthwaite, H.M.; Labine, L.; Watterson, J.H. Semi-quantitative analysis of tramadol, dextromethorphan, and metabolites in decomposed skeletal tissues by ultra performance liquid chromatography quadrupole time of flight mass spectrometry. Drug Test. Anal. 2017, 10, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Gorczynski, L.Y.; Melbye, F.J. Detection of benzodiazepines in different tissues, including bone, using a quantitative ELISA assay. J. Forensic Sci. 2001, 46, 916–918. [Google Scholar] [CrossRef] [PubMed]
- VandenBoer, T.C.; Grummett, S.A.; Watterson, J.H. Utility of immunoassay in drug screening in skeletal tissues: Sampling considerations in detection of ketamine exposure in femoral bone and bone marrow following acute administration using ELISA. J. Forensic Sci. 2008, 53, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Watterson, J.H.; VandenBoer, T.C. Effects of tissue type and the dose-death interval on the detection of acute ketamine exposure in bone marrow with solid-phase extraction and ELISA with liquid chromatography-tandem mass spectrometry confirmation. J. Anal. Toxicol. 2008, 32, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Watterson, J.H.; Botman, J.E. Detection of acute diazepam exposure in bone and marrow: Influence of tissue type and the dose-death interval on sensitivity of detection by ELISA with liquid chromatography tandem mass spectrometry confirmation. J. Forensic Sci. 2009, 54, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Lafreniere, N.M.; Watterson, J.H. Detection of acute fentanyl exposure in fresh and decomposed skeletal tissues. Part II: The effect of dose-death interval. Forensic Sci. Int. 2010, 94, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Cornthwaite, H.M.; Watterson, J.H. The influence of body position and microclimate on ketamine and metabolite distribution in decomposed skeletal remains. J. Anal. Toxicol. 2014, 38, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Unger, K.A.; Watterson, J.H. Analysis of dextromethorphan and dextrorphan in skeletal tissues following decomposition in different microclimates condition. J. Anal. Toxicol. 2016, 40, 669–676. [Google Scholar] [CrossRef]
- Lafreniere, N.M.; Watterson, J.H. Detection of acute fentanyl exposure in fresh and decomposed skeletal tissues. Forensic Sci. Int. 2009, 185, 100–106. [Google Scholar] [CrossRef]
- Ceigniz, S.; Ulukan, O.; Ates, I.; Tugcu, H. Determination of morphine in postmortem rabbit bone marrow and comparison with blood morphine concentrations. Forensic Sci. Int. 2006, 156, 91–94. [Google Scholar]
- Horak, E.L.; Jenkins, A.J. Postmortem tissue distribution of olanzapine and citalopram in a drug intoxication. J. Forensic Sci. 2005, 50, 1–3. [Google Scholar] [CrossRef]
- McIntyre, I.M.; King, C.V.; Boratto, M.; Drummer, O.H. Postmortem drug analyses in bone and bone marrow. Ther. Drug Monit. 2000, 22, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Orfanidis, A.; Gika, H.; Mastrogianni, O.; Krokos, A.; Theodoridis, G.; Zaggelidou, E.; Raikos, N. Determination of drugs of abuse and pharmaceuticals in skeletal tissue by UHPLC–MS/MS. Forensic Sci. Int. 2018, 290, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Vardakou, I.; Athanaselis, S.; Pistos, C.; Papadodima, S.; Spiliopoulou, C.; Moraitis, K. The clavicle bone as an alternative matrix in forensic toxicological analysis. J. Forensic Leg. Med. 2014, 22, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Kudo, K.; Sugie, H.; Syoui, N.; Kurihara, K.; Jitsufuchi, N.; Imamura, T.; Ikeda, N. Detection of triazolam in skeletal remains buried for 4 years. Int. J. Leg. Med. 1997, 110, 281–283. [Google Scholar] [CrossRef]
- Orfanidis, A.; Gika, H.; Zaggelidou, E.; Mastrogianni, O.; Raikos, N. Alprazolam and Zolpidem in Skeletal Tissue of Decomposed Body Confirms Exposure. J. Forensic Sci. 2019, 64, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lopez, L.; Luna-Maldonado, A.; Falcon, M.; Mastrobattista, L.; Navarro-Zaragoza, J.; Mancini, R. Development and validation of a gas chromatography–mass spectrometry method for opiates and cocaine in human bone. J. Pharm. Biomed. Anal. 2019, 164, 636–641. [Google Scholar] [CrossRef]
- Fernandez-Lopez, L.; Pellegrini, M.; Rotolo, M.C.; Luna, A.; Falcon, M.; Mancini, R. Development and validation of a method for analysing of duloxetine, venlafaxine and amitriptyline in human bone. Forensic Sci. Int. 2019, 299, 154–160. [Google Scholar] [CrossRef]
- World Health Organization. The Global Burden of Disease: 2004 Update. Geneva (2008). Available online: https://www.who.int/healthinfo/global_burden_disease/2004_report_update/en/ (accessed on 2 May 2019).
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; A AlMazroa, M.; Amann, M.; Anderson, H.R.; Andrews, K.G.; et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef]
- Johnson, R.D.; Russell, J.L. Quantitation of atenolol, metoprolol, and propranolol in postmortem human fluid and tissue specimens via LC/APCI-MS. Forensic Sci. Int. 2006, 156, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Bangalore, S.; Messerli, F.H.; Kostis, J.B.; Pepine, C.J. Cardiovascular protection using beta-blockers: A critical review of the evidence. J. Am. Coll. Cardiol. 2007, 50, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Grassi, G. The European Society of Cardiology (ESC)/European Society of Hypertension (ESH) 2018 guidelines for hypertension diagnosis and treatment: New concepts and recommendations. Pharmacol. Res. 2019, 139, 489–490. [Google Scholar] [CrossRef] [PubMed]
- Chobanian, A.V.; Bakris, G.L.; Black, H.R.; Cushman, W.C.; Green, L.A.; Izzo, J.J.L.; Jones, D.W.; Materson, B.J.; Oparil, S.; Wright, J.J.T.; et al. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: The JNC 7 report. JAMA 2003, 289, 2560–2572. [Google Scholar] [CrossRef] [PubMed]
- Freemantle, N.; Cleland, J.; Young, P.; Mason, J.; Harrison, J. Beta Blockade after myocardial infarction: Systematic review and meta regression analysis. BMJ 1999, 318, 1730–1737. [Google Scholar] [CrossRef] [PubMed]
- Brophy, J.M.; Joseph, L.; Rouleau, J.L. Beta-blockers in congestive heart failure. A Bayesian meta-analysis. Ann. Intern. Med. 2001, 134, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Mangano, D.T.; Layug, E.L.; Wallace, A.; Tateo, I. Atenolol reduced mortality and cardiovascular events after noncardiac surgery. N. Engl. J. Med. 1996, 335, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Newman, W.J.; Xiong, G.L.; Barnhorst, A.V. Beta-Blockers: Off-Label Use in Psychiatric Disorders. Psychopharm Rev. 2013, 48, 73–80. [Google Scholar] [CrossRef]
- World Anti-Doping Agency. Prohibited List. The World Anti-Doping Code International Standard. 2019. Available online: https://www.wada-ama.org/ (accessed on 2 May 2019).
- Wiysonge, C.S.; Bradley, H.A.; Volmink, J.; Mayosi, B.M.; Opie, L.H. Beta-blockers for hypertension. Cochrane Database Syst. Rev. 2017, 1. [Google Scholar] [CrossRef] [PubMed]
- Berenson, A. Big drug makers see sales decline with their image. The New York Times, 14 November 2005. [Google Scholar]
- Agencia Española de Medicamentos y Productos Sanitarios. Observatorio de uso d Medicamentos. Utilización de Medicamentos Antihipertensivos en España Durante el Periodo 2010–2017. Available online: https://www.aemps.gob.es/en/medicamentosUsoHumano/observatorio/informes-publicados/informes-antihipertensivos-espana-2010-2017.htm (accessed on 2 May 2019).
- Gummin, D.D.; Mowry, J.B.; Spyker, D.A.; Brooks, D.E.; Osterthaler, K.M.; Banner, W. 2017 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 35th Annual Report. Clin. Toxicol. 2018, 56, 1213–1415. [Google Scholar] [CrossRef] [PubMed]
- Mozayani, A.; Singer, P.; Jones, G. Distribution of metoprolol enantiomers in a fatal overdose. J. Anal. Toxicol. 1995, 19, 519–521. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, H.; Taniguchi, T.; Nishiguchi, M.; Ouchi, H.; Minami, T.; Utsumi, T.; Motomura, H.; Tsuda, T.; Ohta, T.; Aoki, S.; et al. An autopsy case of combined drug intoxication involving verapamil, metoprolol and digoxin. Forensic Sci. Int. 2003, 133, 107–112. [Google Scholar] [CrossRef]
- Fucci, N.; Offidani, C. An unusual death by propranolol ingestion. Am. J. Forensic Med. Pathol. 2000, 21, 56–58. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, R.S.; Cole, S.; Knaster, H.B.; Dahlbert, T. Beta blocker overdose with propranolol and with atenolol. Ann. Emerg. Med. 1985, 14, 161–163. [Google Scholar] [CrossRef]
- Buajordet, I.; Ebbesen, J.; Erikssen, J.; Brørs, O.; Hilberg, T. Fatal adverse drug events: The paradox of drug treatment. J. Intern. Med. 2001, 250, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Schulz, M.; Schmoldt, A. Therapeutic and toxic blood concentrations of more than 800 drugs and other xenobiotics. Pharmazie 2003, 58, 447–474. [Google Scholar] [PubMed]
- Canfield, D.V.; Dubowski, K.M.; Whinnery, J.M.; Forster, E.M. Pilot-reported beta-blockers identified by forensic toxicology analysis of postmortem specimens. J. Anal. Toxicol. 2017, 42, 1–5. [Google Scholar] [CrossRef]
- Krumbiegel, F.; Hastedt, M.; Tsokos, M. Nails are a potential alternative matrix to hair for drug analysis in general unknown screenings by liquid-chromatography quadrupole time-of-flight mass spectrometry. Forensic Sci. Med. Pathol. 2014, 10, 496–503. [Google Scholar] [CrossRef]
- Launiainen, T.; Ojanperä, I. Drug concentrations in post-mortem femoral blood compared with therapeutic concentrations in plasma. Drug Test. Anal. 2014, 6, 308–316. [Google Scholar] [CrossRef]
- Kristoffersen, L.; Øiestad, E.L.; Opdal, M.S.; Krogh, M.; Lundanes, E.; Christophersen, A.S. Simultaneous determination of 6 beta-blockers, 3 calcium-channel antagonists, 4 angiotensin-II antagonists and 1 antiarrhytmic drug in post-mortem whole blood by automated solid phase extraction and liquid chromatography mass spectrometry: Method development and robustness testing by experimental design. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 850, 147–160. [Google Scholar]
- Jankovic, S.M. Pharmacokinetics of selective β1-adrenergic blocking agents: Prescribing implications. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B. Normal bone anatomy and physiology. Clin. J. Am. Soc. Nephrol. 2008, 3, S131–S139. [Google Scholar] [CrossRef] [PubMed]
- Wille, S.M.R.; Coucke, W.; De Baere, T.; Peters, F.T. Update of Standard Practices for New Method Validation in Forensic Toxicology. Curr. Pharm. Des. 2017, 23, 5442–5454. [Google Scholar] [CrossRef] [PubMed]
- Peters, F.T.; Wissenbach, D.K.; Busardò, F.P.; Marchei, E.; Pichini, S. Method Development in Forensic Toxicology. Curr. Pharm. Des. 2017, 23, 5455–5467. [Google Scholar] [CrossRef] [PubMed]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Analyte | Calibration Parameters | LODb (ng/mg) | LOQb (ng/mg) | |
|---|---|---|---|---|
| Equationa | Determination Coefficients (r2) a | |||
| Atenolol | Y = 0.0368 x | 0.997 | 0.1 | 0.1 |
| Bisoprolol | Y = 0.0168 x | 0.999 | 0.3 | 0.0 |
| Analyte | Intra-Assay Precision (RSD) | Intra-Assay Accuracy (Absolute %Error) | Inter-Assay Precision (RSD) | Inter-Assay Accuracy (Absolute %Error) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| QCL* | QCM** | QCH*** | QCL | QCM | QCH | QCL | QCM | QCH | QCL | QCM | QCH | |
| Atenolol | 5 | 5.4 | 2.1 | 7.09 | 0.95 | 1.36 | 5.3 | 5.7 | 2.1 | 11.89 | 6.90 | 2.03 |
| Bisoprolol | 2.5 | 1.6 | 1.7 | 9.39 | 6.06 | 1.66 | 1.5 | 0.3 | 2.2 | 0.94 | 2.71 | 0.16 |
| Substance | RT (min) | Characteristic Mass Fragments (m/z) |
|---|---|---|
| Atenolol | 8.7 | 72-223-294-395 |
| Bisoprolol | 8.3 | 73–107–116-281 |
| Lobivolol | 15.1 | 73-294-549-534 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez-Lopez, L.; Pellegrini, M.; Rotolo, M.C.; Luna, A.; Falcon, M.; Mancini, R. Development and Validation of a Method for the Analysis of Bisoprolol and Atenolol in Human Bone. Molecules 2019, 24, 2400. https://doi.org/10.3390/molecules24132400
Fernandez-Lopez L, Pellegrini M, Rotolo MC, Luna A, Falcon M, Mancini R. Development and Validation of a Method for the Analysis of Bisoprolol and Atenolol in Human Bone. Molecules. 2019; 24(13):2400. https://doi.org/10.3390/molecules24132400
Chicago/Turabian StyleFernandez-Lopez, Lucia, Manuela Pellegrini, Maria Concetta Rotolo, Aurelio Luna, Maria Falcon, and Rosanna Mancini. 2019. "Development and Validation of a Method for the Analysis of Bisoprolol and Atenolol in Human Bone" Molecules 24, no. 13: 2400. https://doi.org/10.3390/molecules24132400