Molecular Informatics Studies of the Iron-Dependent Regulator (ideR) Reveal Potential Novel Anti-Mycobacterium ulcerans Natural Product-Derived Compounds

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Three Dimensional (3D) Model Prediction and Validation
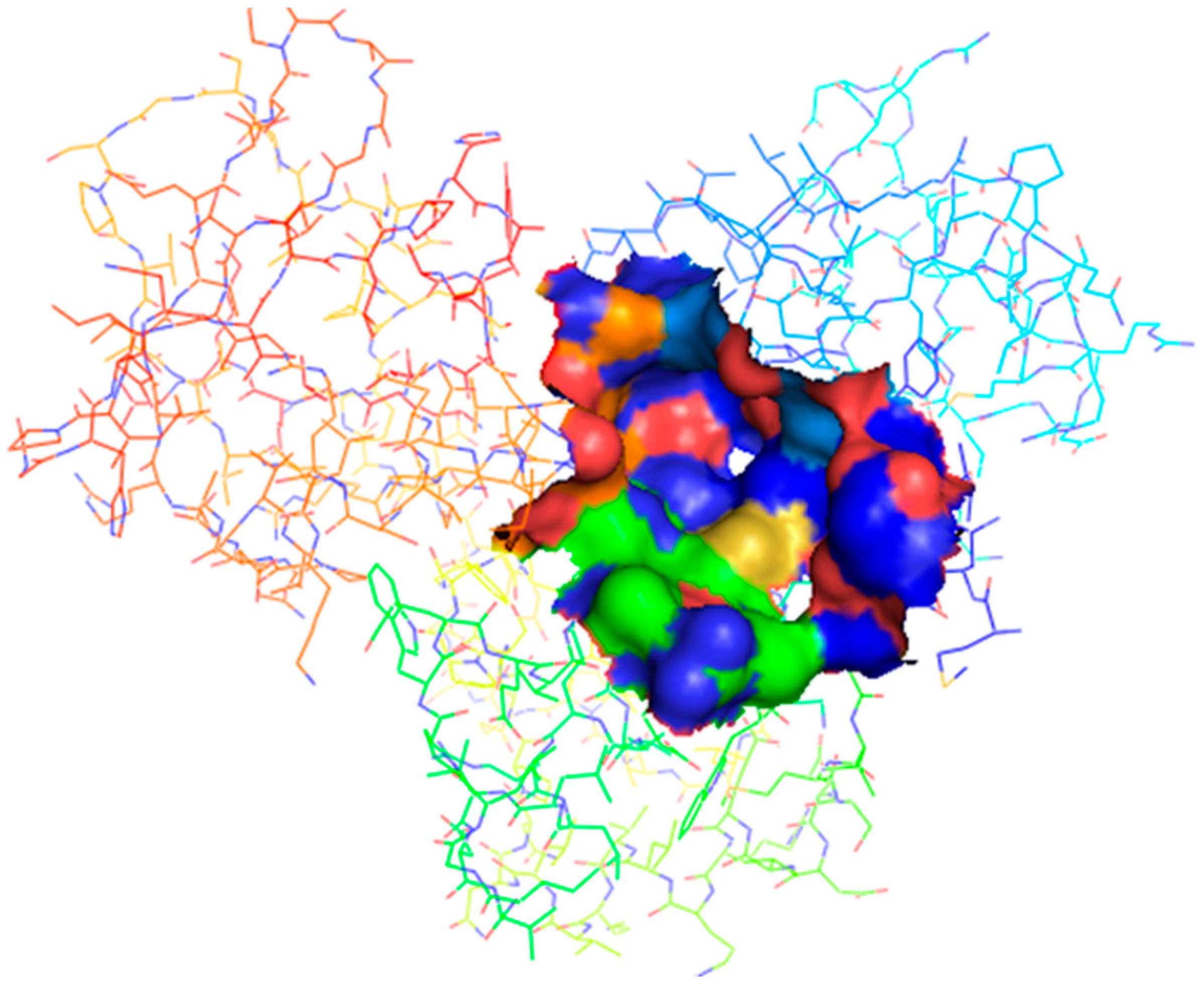
2.2. Binding Site Identification
2.3. Anti-Mycobacterial Lead Discovery
2.4. Evaluation of Autodock Vina’s Performance
2.5. In Silico ADMET Studies
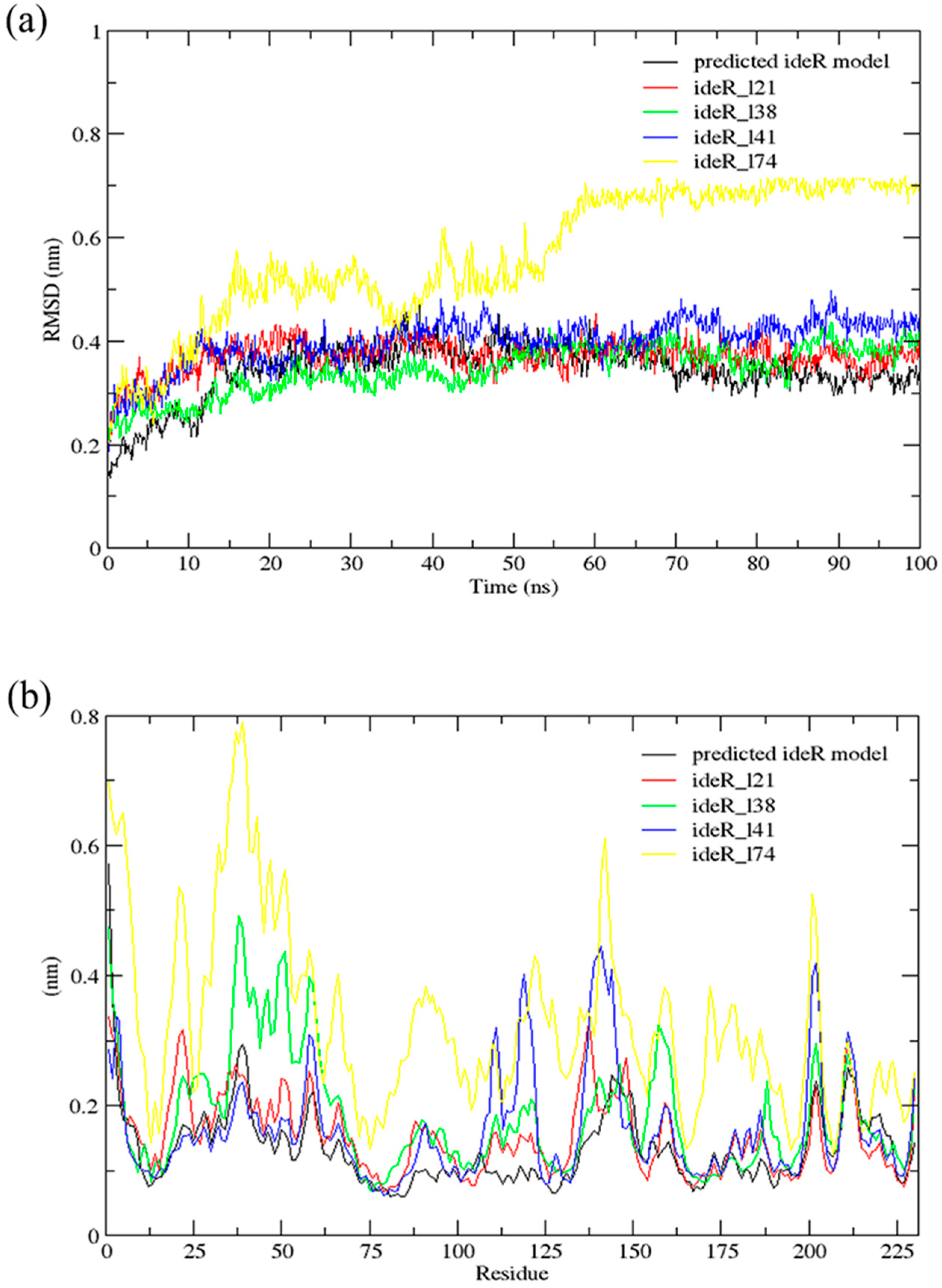
2.6. Molecular Dynamics (MD) Simulations
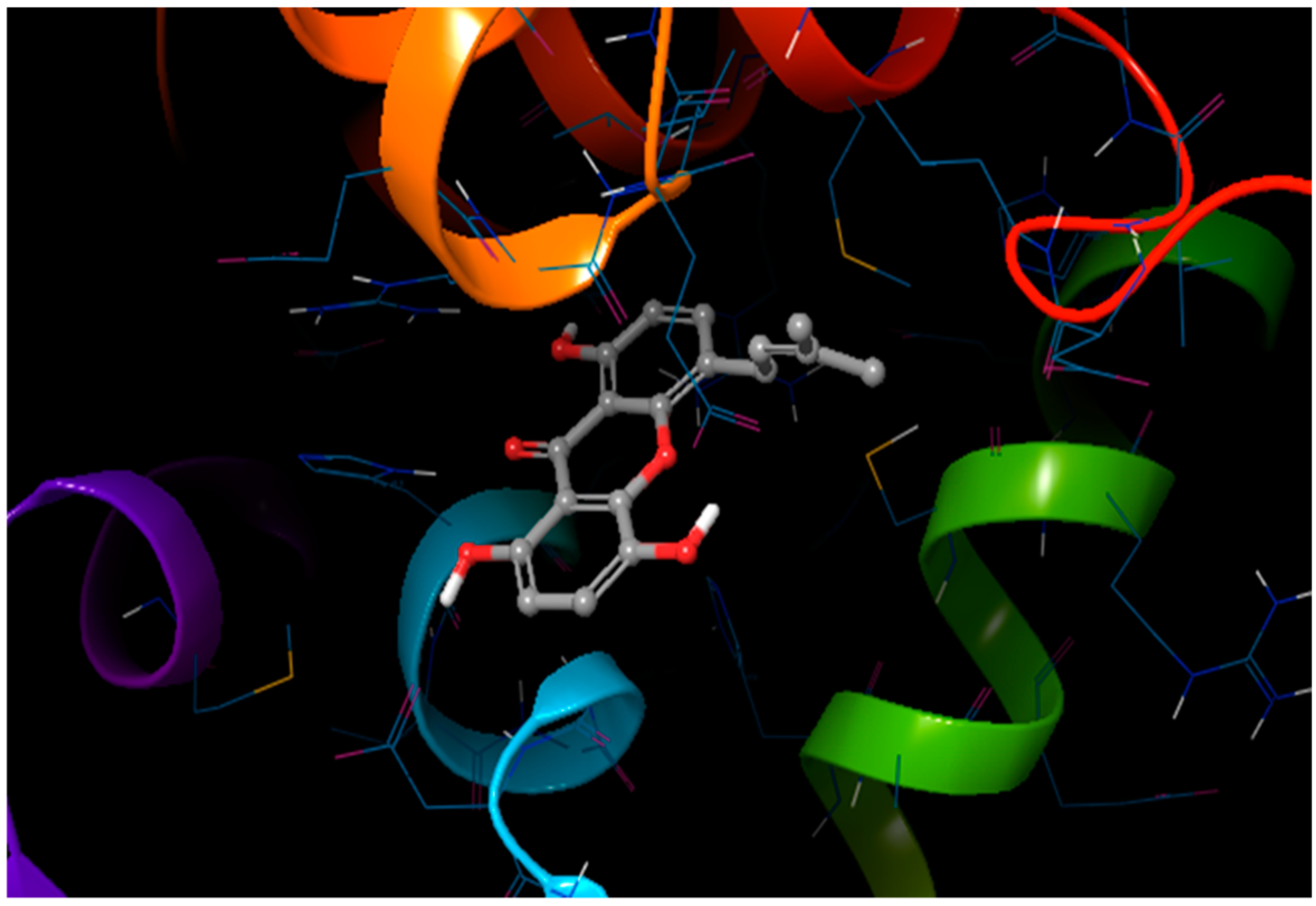
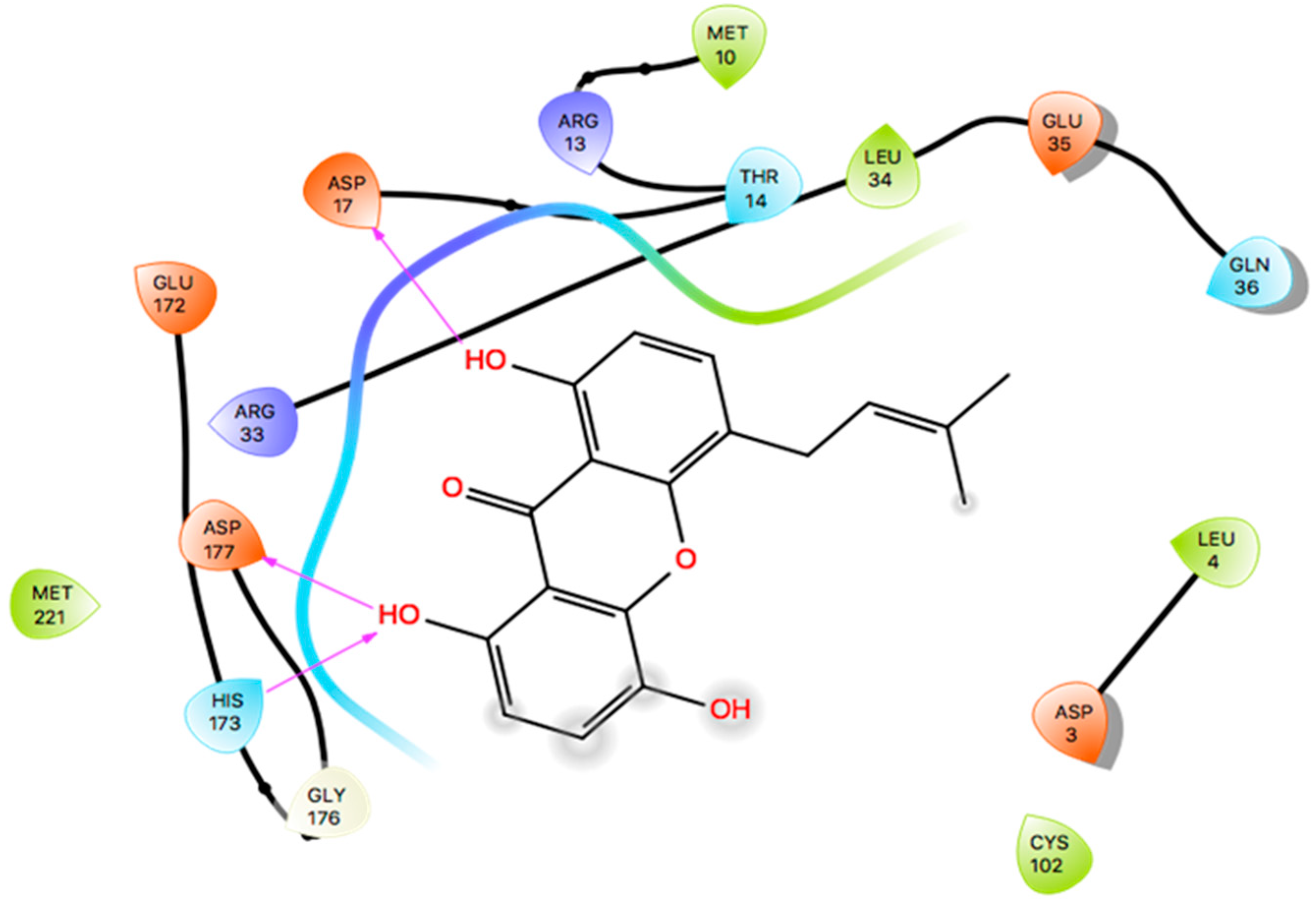
2.7. Induced Fit Docking
3. Screening of Lead Compounds and Known Inhibitors against the DNA-Binding Site
4. Exploring the Anti-Mycobacterial Activity of the Predicted Leads
5. Materials and Methods
5.1. Homology Modeling of Mycobacterium ulcerans ideR Structure
5.2. Structure Validation
5.3. Binding Site (Pocket) Identification
5.4. Virtual Screening
5.5. Validation of Docking Protocol
5.6. In Silico ADMET Studies
5.7. Molecular Dynamic Simulations
5.8. Induced Fit Docking
5.9. Lead Structural Similarity Searches and Antimycobacterial Association
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nyabadza, F.; Bonyah, E. On the transmission dynamics of Buruli ulcer in Ghana: Insights through a mathematical model. BMC Res. Notes 2015, 8, 656. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.S.; Hill, K.; McKenna, M.; Ogbechi, J.; High, S.; Willis, A.E.; Simmonds, R.E. The Pathogenic Mechanism of the Mycobacterium ulcerans Virulence Factor, Mycolactone, Depends on Blockade of Protein Translocation into the ER. PLoS Pathog. 2014, 10, e1004061. [Google Scholar] [CrossRef] [PubMed]
- Williamson, H.R.; Benbow, M.E.; Nguyen, K.D.; Beachboard, D.C.; Kimbirauskas, R.K.; McIntosh, M.D.; Quaye, C.; Ampadu, E.O.; Boakye, D.; Merritt, R.W.; et al. Distribution of Mycobacterium ulcerans in Buruli Ulcer Endemic and Non-Endemic Aquatic Sites in Ghana. PLoS Negl. Trop. Dis. 2008, 2, e205. [Google Scholar] [CrossRef] [PubMed]
- CDC. CDC—Neglected Tropical Diseases—Diseases. Available online: http://www.cdc.gov/globalhealth/ntd/diseases/index.html (accessed on 18 January 2018).
- WHO. WHO|Buruli Ulcer. Available online: http://www.who.int/buruli/en/ (accessed on 17 February 2017).
- Ampah, K.A.; Asare, P.; Binnah, D.D.-G.; Maccaulley, S.; Opare, W.; Röltgen, K.; Pluschke, G.; Yeboah-Manu, D. Burden and Historical Trend of Buruli Ulcer Prevalence in Selected Communities along the Offin River of Ghana. PLoS Negl. Trop. Dis. 2016, 10, e0004603. [Google Scholar] [CrossRef] [PubMed]
- Anand, U.; Sinisi, M.; Fox, M.; MacQuillan, A.; Quick, T.; Korchev, Y.; Bountra, C.; McCarthy, T.; Anand, P. Mycolactone-mediated neurite degeneration and functional effects in cultured human and rat DRG neurons: Mechanisms underlying hypoalgesia in Buruli ulcer. Mol. Pain 2016, 12, 174480691665414. [Google Scholar] [CrossRef] [PubMed]
- Garchitorena, A.; Guégan, J.-F.; Léger, L.; Eyangoh, S.; Marsollier, L.; Roche, B. Mycobacterium ulcerans dynamics in aquatic ecosystems are driven by a complex interplay of abiotic and biotic factors. eLife 2015, 4, e07616. [Google Scholar] [CrossRef]
- Amofah, G.; Bonsu, F.; Tetteh, C.; Okrah, J.; Asamoa, K.; Asiedu, K.; Addy, J. Buruli ulcer in Ghana: Results of a national case search. Emerg. Infect. Dis. 2002, 8, 167–170. [Google Scholar] [CrossRef]
- Mosi, L.; Williamson, H.; Wallace, J.R.; Merritt, R.W.; Small, P.L.C. Persistent association of Mycobacterium ulcerans with West African predaceous insects of the family belostomatidae. Appl. Environ. Microbiol. 2008, 74, 7036–7042. [Google Scholar] [CrossRef]
- Azumah, B.K.; Addo, P.G.; Dodoo, A.; Awandare, G.; Mosi, L.; Boakye, D.A.; Wilson, M.D. Experimental demonstration of the possible role of Acanthamoeba polyphaga in the infection and disease progression in Buruli Ulcer (BU) using ICR mice. PLoS ONE 2017, 12, e0172843. [Google Scholar] [CrossRef]
- Bieri, R.; Pluschke, G.; Huber, S.; Li, J.; Scherr, N.; Bomio, C.; Hug, M.N.; Gersbach, P.; Altmann, K.-H.; Dangy, J.-P. Antibody-Mediated Neutralization of the Exotoxin Mycolactone, the Main Virulence Factor Produced by Mycobacterium ulcerans. PLoS Negl. Trop. Dis. 2016, 10, e0004808. [Google Scholar]
- Adusumilli, S. Understanding Immune Response in Mycobacterium ulcerans Infection. Ph.D. Thesis, University of Tennessee, Knoxville, TN, USA, 2005. Available online: https://trace.tennessee.edu/utk_graddiss/656 (accessed on 1 April 2019).
- Kwofie, S.; Dankwa, B.; Enninful, K.; Adobor, C.; Broni, E.; Ntiamoah, A.; Wilson, M. Molecular Docking and Dynamics Simulation Studies Predict Munc18b as a Target of Mycolactone: A Plausible Mechanism for Granule Exocytosis Impairment in Buruli Ulcer Pathogenesis. Toxins 2019, 11, 181. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Chandra, N.; Vishveshwara, S. Mechanism of Iron-Dependent Repressor (IdeR) Activation and DNA Binding: A Molecular Dynamics and Protein Structure Network Study. PLoS Comput. Biol. 2015, 11, e1004500. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, S.; Yellaboina, S.; Ranjan, A. IdeR in mycobacteria: From target recognition to physiological function. Crit. Rev. Microbiol. 2006, 32, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Deshayes, C.; Angala, S.K.; Marion, E.; Brandli, I.; Babonneau, J.; Preisser, L.; Eyangoh, S.; Delneste, Y.; Legras, P.; De Chastellier, C.; et al. Regulation of Mycolactone, the Mycobacterium ulcerans Toxin, Depends on Nutrient Source. PLoS Negl. Trop. Dis. 2013, 7, e2502. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, A.A.; Iram, F.; Siddiqui, S.; Sahu, K. Role of natural products in drug discovery process. Int. J. Drug Dev. Res. 2014, 6, 172–204. [Google Scholar]
- Altschul, S. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Moorhouse, M.; Barry, P. The Protein Databank. In Bioinformatics Biocomputing and Perl; John Wiley & Sons, Ltd.: Chichester, UK, 2005; pp. 173–210. [Google Scholar]
- Kuntal, B.K.; Aparoy, P.; Reddanna, P. EasyModeller: A graphical interface to MODELLER. BMC Res. Notes 2010, 3, 226. [Google Scholar] [CrossRef]
- Shen, M.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [Green Version]
- Vriend, G. WHAT IF: A molecular modeling and drug design program. J. Mol. Graph. 1990, 8, 52–56. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 2002, 26, 283–291. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Yang, J.; Zhang, Y. COFACTOR: An accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 2012, 40, W471–W477. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. BioLiP: A semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res. 2013, 41, D1096–D1103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins Struct. Funct. Genet. 2004, 57, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Goranson-Siekierke, J.; Ingason, B.P.; Holmes, R.K.; Hol, W.G.J.; Feese, M.D. Crystal Structure of the Iron-dependent Regulator from Mycobacterium tuberculosis at 2.0-Å Resolution Reveals the Src Homology Domain 3-like Fold and Metal Binding Function of the Third Domain. J. Biol. Chem. 2002, 276, 5959–5966. [Google Scholar]
- Rohilla, A.; Khare, G.; Tyagi, A.K. Virtual Screening, pharmacophore development and structure based similarity search to identify inhibitors against IdeR, a transcription factor of Mycobacterium tuberculosis. Sci. Rep. 2017, 7, 4653. [Google Scholar] [CrossRef]
- Ntie-Kang, F.; Zofou, D.; Babiaka, S.B.; Meudom, R.; Scharfe, M.; Lifongo, L.L.; Mbah, J.A.; Mbaze, L.M.A.; Sippl, W.; Efange, S.M.N. AfroDb: A select highly potent and diverse natural product library from African medicinal plants. PLoS ONE 2013, 8, e78085. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Truchon, J.-F.; Bayly, C.I. Evaluating Virtual Screening Methods: Good and Bad Metrics for the “Early Recognition” Problem. J. Chem. Inf. Model. 2007, 47, 488–508. [Google Scholar] [CrossRef]
- Goksuluk, D.; Korkmaz, S.; Zararsiz, G.; Karaagaoglu, A.E. EasyROC: An interactive web-tool for roc curve analysis using r language environment. R J. 2016, 8, 213–230. [Google Scholar] [CrossRef]
- Triballeau, N.; Acher, F.; Brabet, I.; Pin, J.P.; Bertrand, H.O. Virtual screening workflow development guided by the “receiver operating characteristic” curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4. J. Med. Chem. 2005, 48, 2534–2547. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, J.N. Receiver operating characteristic curve in diagnostic test assessment. J. Thorac. Oncol. 2010, 5, 1315–1316. [Google Scholar] [CrossRef]
- Kwofie, S.; Dankwa, B.; Odame, E.; Agamah, F.; Doe, L.; Teye, J.; Agyapong, O.; Miller, W.; Mosi, L.; Wilson, M. In Silico Screening of Isocitrate Lyase for Novel Anti-Buruli Ulcer Natural Products Originating from Africa. Molecules 2018, 23, 1550. [Google Scholar] [CrossRef] [PubMed]
- Shamsara, J. Correlation between Virtual Screening Performance and Binding Site Descriptors of Protein Targets. Int. J. Med. Chem. 2018, 2018, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiroga, R.; Villarreal, M.A. Vinardo: A scoring function based on autodock vina improves scoring, docking, and virtual screening. PLoS ONE 2016, 11, e0155183. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Sperandio, O.; Galons, H.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs2: Free ADME/tox filtering tool to assist drug discovery and chemical biology projects. BMC Bioinform. 2008, 9, 396. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- Kubinyi, H. Hydrogen Bonding: The Last Mystery in Drug Design? In Pharmacokinetic Optimization in Drug Research; Verlag Helvetica Chimica Acta: Zürich, Switzerland, 2007; pp. 513–524. [Google Scholar]
- Benigni, R.; Bossa, C. Structural Alerts of Mutagens and Carcinogens. Curr. Comput. Aided-Drug Des. 2006, 2, 169–176. [Google Scholar] [CrossRef]
- Greene, N.; Judson, P.N.; Langowski, J.J.; Marchant, C.A. Knowledge-Based Expert Systems for Toxicity and Metabolism Prediction: DEREK, StAR and METEOR. Sar Qsar Environ. Res. 1999, 10, 299–314. [Google Scholar] [CrossRef]
- Dobo, K.L.; Greene, N.; Fred, C.; Glowienke, S.; Harvey, J.S.; Hasselgren, C.; Jolly, R.; Kenyon, M.O.; Munzner, J.B.; Muster, W.; et al. In silico methods combined with expert knowledge rule out mutagenic potential of pharmaceutical impurities: An industry survey. Regul. Toxicol. Pharmacol. 2012, 62, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Hess, B.; Lindahl, E.; Van Der Spoel, D.; Mark, A.E.; Groenhof, G. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar]
- Tiburu, E.K.; Issah, I.; Darko, M.; Armah-Sekum, R.E.; Gyampo, S.O.A.; Amoateng, N.K.; Kwofie, S.K.; Awandare, G. Investigating the Conformation of S100β Protein Under Physiological Parameters Using Computational Modeling: A Clue for Rational Drug Design. Open Biomed. Eng. J. 2018, 12, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Lill, M.A. Induced fit docking, and the use of QM/MM methods in docking. Drug Discov. Today Technol. 2013, 10, e411–e418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farid, R.; Day, T.; Friesner, R.A.; Pearlstein, R.A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorganic Med. Chem. 2006, 14, 3160–3173. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Beard, H.S.; Farid, R. Use of an induced fit receptor structure in virtual screening. Chem. Biol. Drug Des. 2006, 67, 83–84. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein− ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; Méndez-Lucio, O.; Yoo, J. Rationalization of activity cliffs of a sulfonamide inhibitor of DNA methyltransferases with induced-fit docking. Int. J. Mol. Sci. 2014, 15, 3253–3261. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Scherr, N.; Pluschke, G.; Panda, M. Comparative Study of Activities of a Diverse Set of Antimycobacterial Agents against Mycobacterium tuberculosis and Mycobacterium ulcerans. Antimicrob. Agents Chemother. 2016, 60, 3132–3137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherr, N.; Bieri, R.; Thomas, S.S.; Chauffour, A.; Kalia, N.P.; Schneide, P.; Ruf, M.-T.; Lamelas, A.; Manimekalai, M.S.S.; Grüber, G.; et al. Targeting the Mycobacterium ulcerans cytochrome bc1:aa3 for the treatment of Buruli ulcer. Nat. Commun. 2018, 9, 5370. [Google Scholar] [CrossRef] [PubMed]
- Araujo, R.C.P.; Neves, F.A.R.; Formagio, A.S.N.; Kassuya, C.A.L.; Stefanello, M.E.A.; Souza, V.V.; Pavan, F.R.; Croda, J. Evaluation of the anti-mycobacterium tuberculosis activity and in vivo acute toxicity of Annona sylvatic. BMC Complement. Altern. Med. 2014, 14, 209. [Google Scholar] [CrossRef] [PubMed]
- Kuete, V.; Ngameni, B.; Simo, C.C.F.; Tankeu, R.K.; Ngadjui, B.T.; Meyer, J.J.M.; Lall, N.; Kuiate, J.R. Antimicrobial activity of the crude extracts and compounds from Ficus chlamydocarpa and Ficus cordata (Moraceae). J. Ethnopharmacol. 2008, 120, 17–24. [Google Scholar] [CrossRef]
- Safwat, N.A.; Kashef, M.T.; Aziz, R.K.; Amer, K.F.; Ramadan, M.A. Quercetin 3-O-glucoside recovered from the wild Egyptian Sahara plant, Euphorbia paralias L., inhibits glutamine synthetase and has antimycobacterial activity. Tuberculosis 2018, 108, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Goud, G.L.; Ramesh, S.; Ashok, D.; Reddy, V.P.; Yogeeswari, P.; Sriram, D.; Saikrishna, B.; Manga, V. Design, synthesis, molecular-docking and antimycobacterial evaluation of some novel 1,2,3-triazolyl xanthenones. MedChemComm 2017, 8, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Jamkhande, P.G.; Pathan, S.K.; Wadher, S.J. In silico PASS analysis and determination of antimycobacterial, antifungal, and antioxidant efficacies of maslinic acid in an extract rich in pentacyclic triterpenoids. Int. J. Mycobacteriol. 2016, 5, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of activity spectra for biologically active substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Lätti, S.; Niinivehmas, S.; Pentikäinen, O.T. Rocker: Open source, easy-to-use tool for AUC and enrichment calculations and ROC visualization. J. Cheminform. 2016, 8, 45. [Google Scholar] [CrossRef]
- Empereur-Mot, C.; Zagury, J.F.; Montes, M. Screening Explorer-An Interactive Tool for the Analysis of Screening Results. J. Chem. Inf. Model. 2016, 56, 2281–2286. [Google Scholar] [CrossRef] [PubMed]
- Schüttelkopf, A.W.; Van Aalten, D.M.F. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hits Description | Query Cover | E Value | Identity | PDB ID |
|---|---|---|---|---|
| Chain A, Crystal Structure of ideR from Mycobacterium tuberculosis | 100% | 8 × 10−154 | 92% | 1FX7_A |
| Chain A, Crystal Structure of a two-domain ideR-DNA complex crystal form I | 64% | 1 × 10−96 | 91% | 2ISZ_A |
| Chain A, ideR from M. tuberculosis | 60% | 2 × 10−95 | 96% | 1B1B_A |
| Chain A, Crystal Structure of the Nickel—activated two-domain Iron—dependent Regulator | 64% | 6 × 10−95 | 91% | 2ISY_A |
| Chain A, Diphtheria Tox Repressor (C102d Mutant) complexed with Nickel and Dtxr consensus binding sequence | 52% | 1 × 10−62 | 80% | 1F5T_A |
| BINDING SITE PREDICTION | RESIDUES AT THE BINDING SITE |
| COFACTOR | His79, Glu 83, His 98, Glu172, Gln175 |
| COACH | His219, His223 |
| REPORTED BINDING SITES OF THE TEMPLATE (1FX7) [29] | |
| 1 (Metal binding site 1) | His79, Glu83, His98, Glu172, Gln175 |
| 2 (Metal binding site 2) | Met10, Cys102, Glu105, His106 |
| 3 | His219, His223 |
| 4 | His212 |
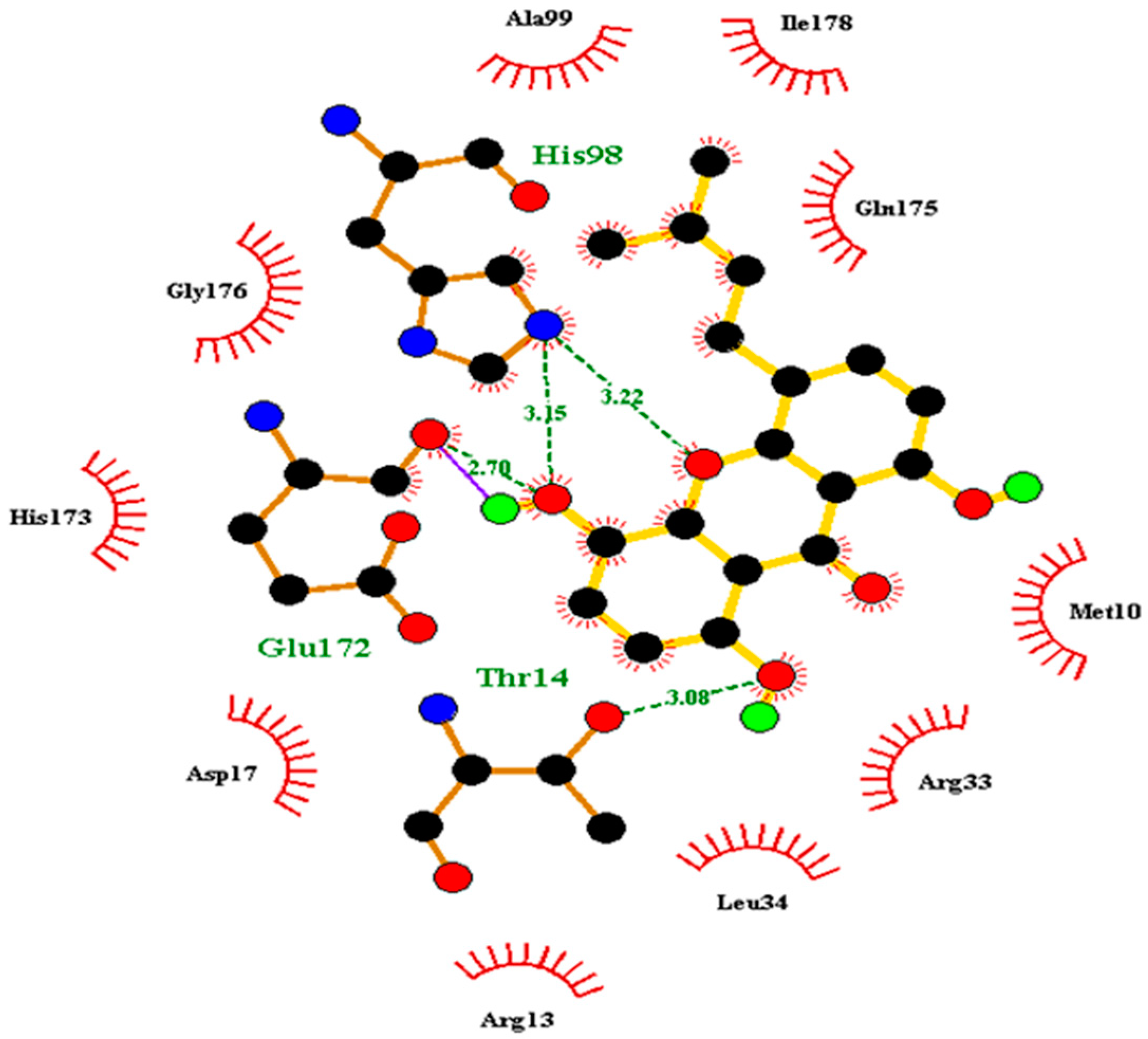
| LIGAND ID | BINDING ENERGY | RESIDUES LIGAND INTERACTS WITH |
|---|---|---|
| NSC12453 | −7.5 | His98 |
| NSC201773 | −7.5 | His98 |
| NSC282699 | −7.5 | His98 |
| NSC303600 | −7 | His98 |
| NSC65748 | −7 | His98 |
| ZINC000005357841 | −7.4 | Cys102, His98, Met10 |
| ZINC000013327497 | −7.1 | Met10, Cys102, His98 |
| ZINC000013481884 | −7.3 | Glu172, His98, Cys102 |
| ZINC000014417338 | −8 | Glu172, His98 |
| ZINC000014811038 | −7.7 | Glu172, His98, Cys102 |
| ZINC000014819573 | −7.4 | His98, Cys102, Met10 |
| ZINC000018185774 | −7.7 | His98, Cys102, Met10 |
| ZINC000033831303 | −7.4 | Met10, Cys102, His98 |
| ZINC000095485893 | −7.2 | Met10, His98, Cys102 |
| ZINC000095485918 | −6.9 | Glu172, Cys102, His98 |
| ZINC000095485921 | −7.6 | Met10, His98, Glu172 |
| ZINC000095486065 | −7.5 | Met10, His98, Cys102, Glu172 |
| ZINC000095486093 | −7.1 | Glu172, Cys102, His98 |
| ZINC000095486151 | −7.3 | Cys102, Met10, His98 |
| ZINC000095486157 | −7 | His98, Met10, Cys102 |
| ZINC000095486193 | −7.2 | Glu172, His98, Cys102 |
| ZINC000095486235 | −8.3 | His98, Cys102 |
| ZINC000095486265 | −7.8 | Met10, His98, Cys102 |
| ZINC000095486301 | −7.6 | His98, Met10, Cys102 |
| ZINC000095486336 | −8.4 | Glu172, His98 |
| ROC_AUC | BEDROC (alpha = 20.0) | Enrichment Factor | |||
|---|---|---|---|---|---|
| 1% | 10% | 20% | |||
| BU_MBS | 0.702 | 0.137 | 2.979 | 2.355 | 2.208 |
| BU_DBS | 0.743 | 0.143 | 2.979 | 2.355 | 2.650 |
| TB_MBS | 0.727 | 0.174 | 0 | 2.355 | 2.797 |
| TB_DBS | 0.703 | 0.175 | 5.95 | 2.650 | 2.355 |
| Ligands | Status |
|---|---|
| ZINC000005357841 | Accepted |
| ZINC000014417338 | Accepted |
| ZINC000018185774 | Intermediate |
| ZINC000095485921 | Intermediate |
| Ligands. | End Point | Species | Reasoning Outcome | Negative Outcome | Strongest Ec3 Prediction |
|---|---|---|---|---|---|
| ZINC000005357841 | Hepatotoxicity | Mammal | Plausible | - | - |
| Mutagenicity In Vitro | Bacterium | - | Inactive | - | |
| Carcinogenicity | Mammal | Plausible | - | - | |
| Skin Sensitization | Mammal | Plausible | - | 2.9% Moderate Sensitizer | |
| ZINC000018185774 | Teratogenicity | Mammal | Equivocal | - | - |
| Mutagenicity In Vitro | Bacterium | - | Inactive | ||
| Skin Sensitization | Mammal | Plausible | - | 0.15% Strong Sensitizer | |
| ZINC000095485921 | Photoallergenicity | Mammal | Plausible | - | - |
| Teratogenicity | Mammal | Equivocal | - | - | |
| Mutagenicity In Vitro | Bacterium | - | Inactive | ||
| Skin Sensitization | Mammal | Plausible | - | 0.15% Strong Sensitizer | |
| ZINC000014417338 | Mutagenicity In Vitro | Bacterium | - | Inactive | |
| Skin Sensitization | Mammal | Plausible | - | 0.16% Strong Sensitizer |
| Ligand ID | Lipinski’s Violation | Solubility (mg/L) | Solubility Forecast Index | Oral Bioavailability (Veber) |
|---|---|---|---|---|
| ZINC000014417338 | 0 | 3279.99 | Reduced Solubility | Good |
| ZINC000005357841 | 0 | 4526.38 | Reduced Solubility | Good |
| ZINC000018185774 | 0 | 8434.39 | Good Solubility | Good |
| ZINC000095485921 | 0 | 2928.86 | Reduced Solubility | Good |
| Moxifloxacin | 0 | 30151.64 | Good Solubility | Good |
| Amikacin | 3 | 5077659.17 | Good Solubility | Low |
| Streptomycin | 3 | 3508974.65 | Good Solubility | Low |
| Clarithromycin | 2 | 1491.87 | Good Solubility | Good |
| Rifampicin | 4 | 246.01 | Good Solubility | Good |
| Ligand ID | Metal Binding Site 2 | DNA-Binding Site | ||
|---|---|---|---|---|
| Binding Energy (KCAL/MOL) | Hydrogen Bonds | Binding Energy (KCAL/MOL) | Hydrogen Bonds | |
| NSC12453 | −7.5 | Gly176, Arg13, His98 | −5.9 | Gln43, Arg47, Thr7 |
| NSC201773 | −7.5 | Gly176, His173, His98 | −6 | Arg27 |
| NSC282699 | −7.5 | - | −5.9 | Arg47, Thr44 |
| NSC303600 | −7 | Thr14, Arg13, Arg33 | −5.9 | Thr8, Thr7, Asn2 |
| NSC65748 | −7 | Arg33, Asp17, His173, Arg13 | −5.5 | Ser37, Thr40, Gln36, Glu35 |
| ZINC000014417338 | −8 | Arg33, Asp17, His98 | −5.9 | Ser42 |
| ZINC000018185774 | −7.7 | Asp3, Arg103, Arg33, Asp17, Glu172 | −5.8 | Arg47, Thr44 |
| ZINC000095485921 | −7.6 | Thr14, His98, Glu172 | −5.7 | Arg60, Ser42, Arg27, Ala28 |
| ZINC000005357841 | −7.4 | His98 | −5.9 | Thr7, Asn2, Thr44 |
| Ligand ID | Common Names | Two-Dimensional Structure |
|---|---|---|
| ZINC000014417338 | Alpinumisoflavone; 5-hydroxy-7-(4-hydroxyphenyl)-2,2-dimethylpyrano[3,2-g]chromen-6-one |  |
| ZINC000005357841 | (6-methoxybenzo[1,3]dioxol-5-yl)BLAHone |  |
| ZINC000018185774 | Luteolin; 2-(3,4-Dihydroxy-phenyl)-5,7-dihydroxy-chromen-4-one |  |
| ZINC000095485921 | 1,4,8-trihydroxy-5-(3-methylbut-2-enyl)xanthen-9-one | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwofie, S.K.; Enninful, K.S.; Yussif, J.A.; Asante, L.A.; Adjei, M.; Kan-Dapaah, K.; Tiburu, E.K.; Mensah, W.A.; Miller, W.A., III; Mosi, L.; et al. Molecular Informatics Studies of the Iron-Dependent Regulator (ideR) Reveal Potential Novel Anti-Mycobacterium ulcerans Natural Product-Derived Compounds. Molecules 2019, 24, 2299. https://doi.org/10.3390/molecules24122299
Kwofie SK, Enninful KS, Yussif JA, Asante LA, Adjei M, Kan-Dapaah K, Tiburu EK, Mensah WA, Miller WA III, Mosi L, et al. Molecular Informatics Studies of the Iron-Dependent Regulator (ideR) Reveal Potential Novel Anti-Mycobacterium ulcerans Natural Product-Derived Compounds. Molecules. 2019; 24(12):2299. https://doi.org/10.3390/molecules24122299
Chicago/Turabian StyleKwofie, Samuel K., Kweku S. Enninful, Jaleel A. Yussif, Lina A. Asante, Mavis Adjei, Kwabena Kan-Dapaah, Elvis K. Tiburu, Wilhelmina A. Mensah, Whelton A. Miller, III, Lydia Mosi, and et al. 2019. "Molecular Informatics Studies of the Iron-Dependent Regulator (ideR) Reveal Potential Novel Anti-Mycobacterium ulcerans Natural Product-Derived Compounds" Molecules 24, no. 12: 2299. https://doi.org/10.3390/molecules24122299