
Dioscin Inhibits the Invasion and Migration of Hepatocellular Carcinoma HepG2 Cells by Reversing TGF-β1-Induced Epithelial-Mesenchymal Transition

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
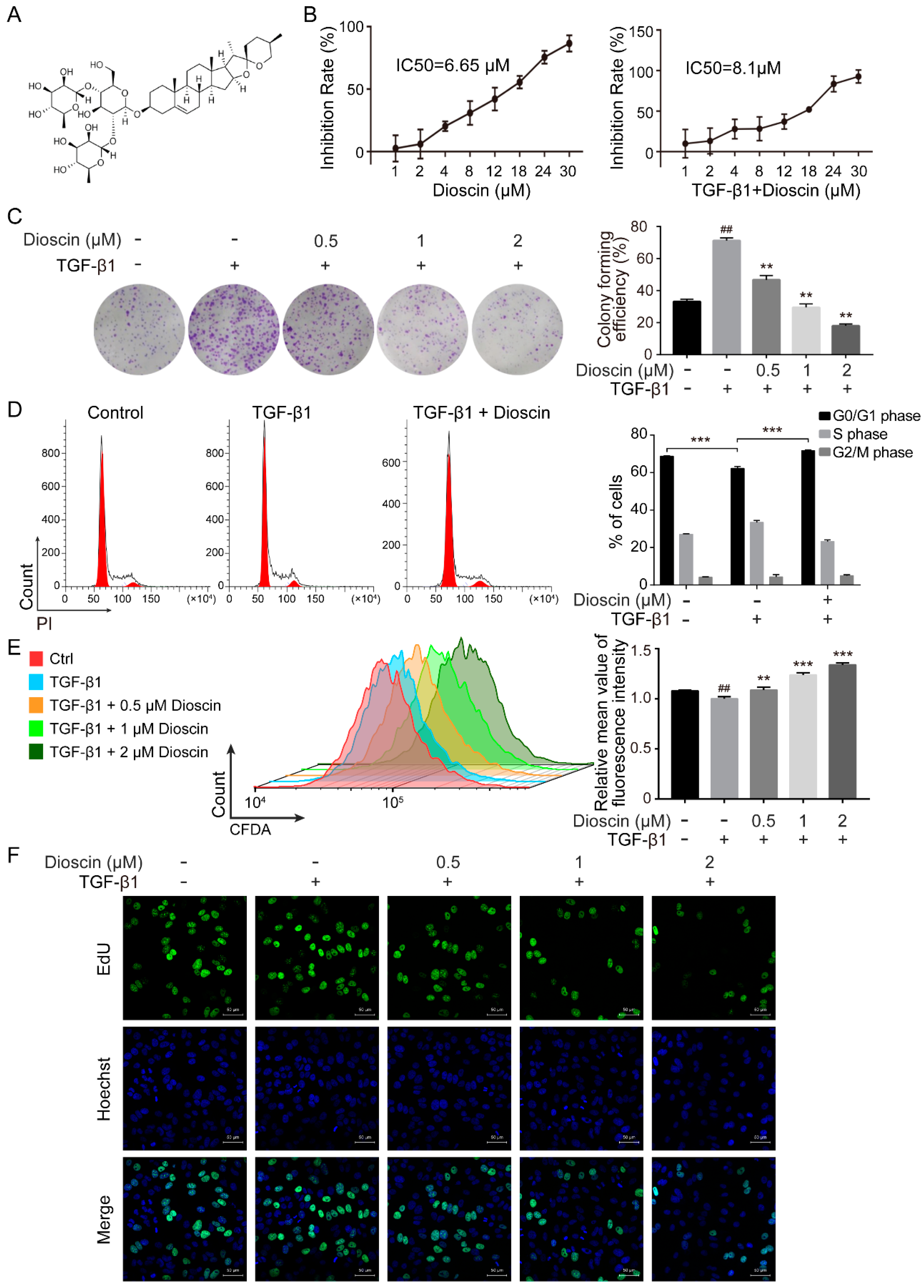
2.1. Dioscin Inhibits Proliferation of HepG2 Cells
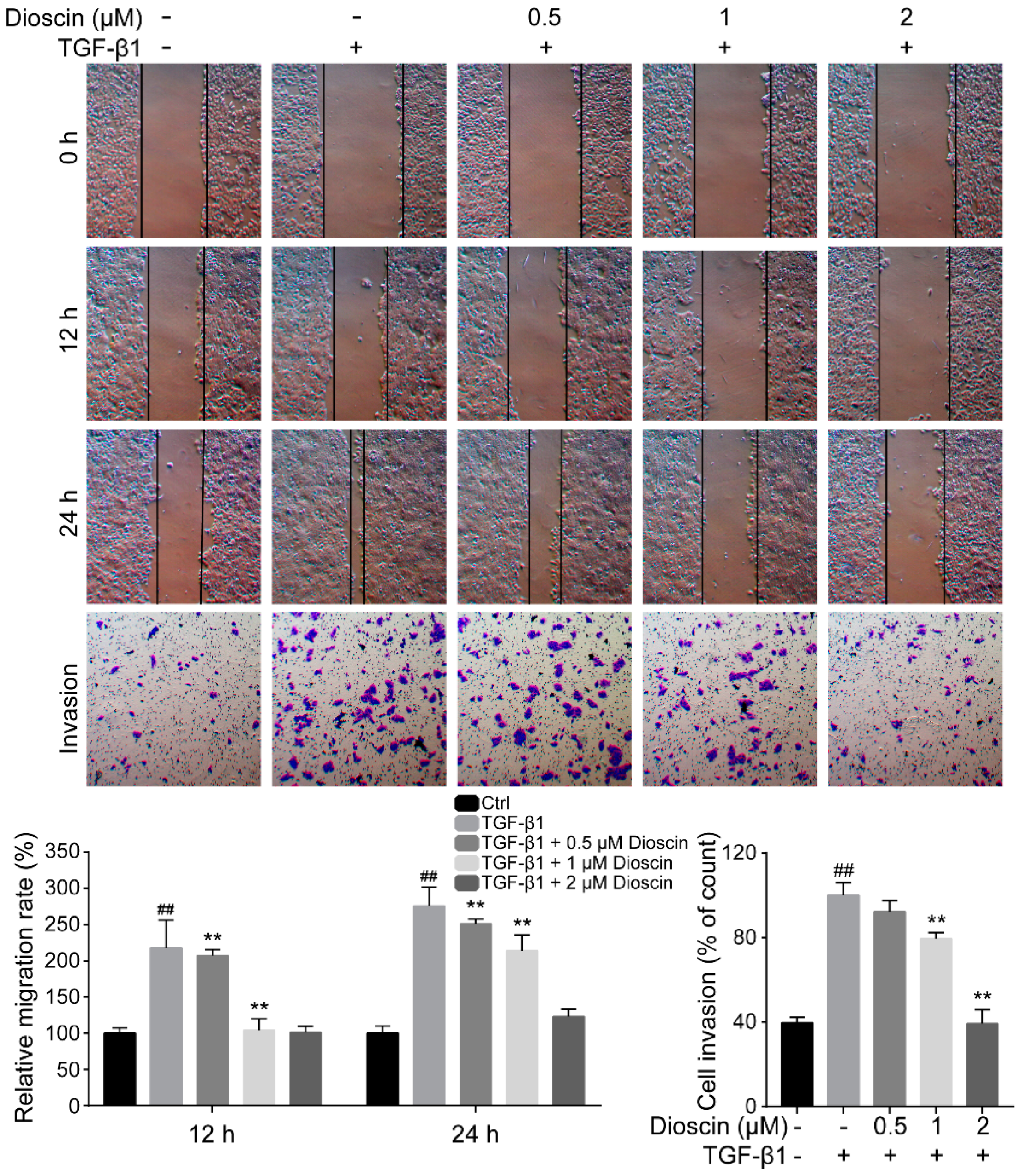
2.2. Dioscin Inhibits the Migration and Invasion of HepG2 Cells
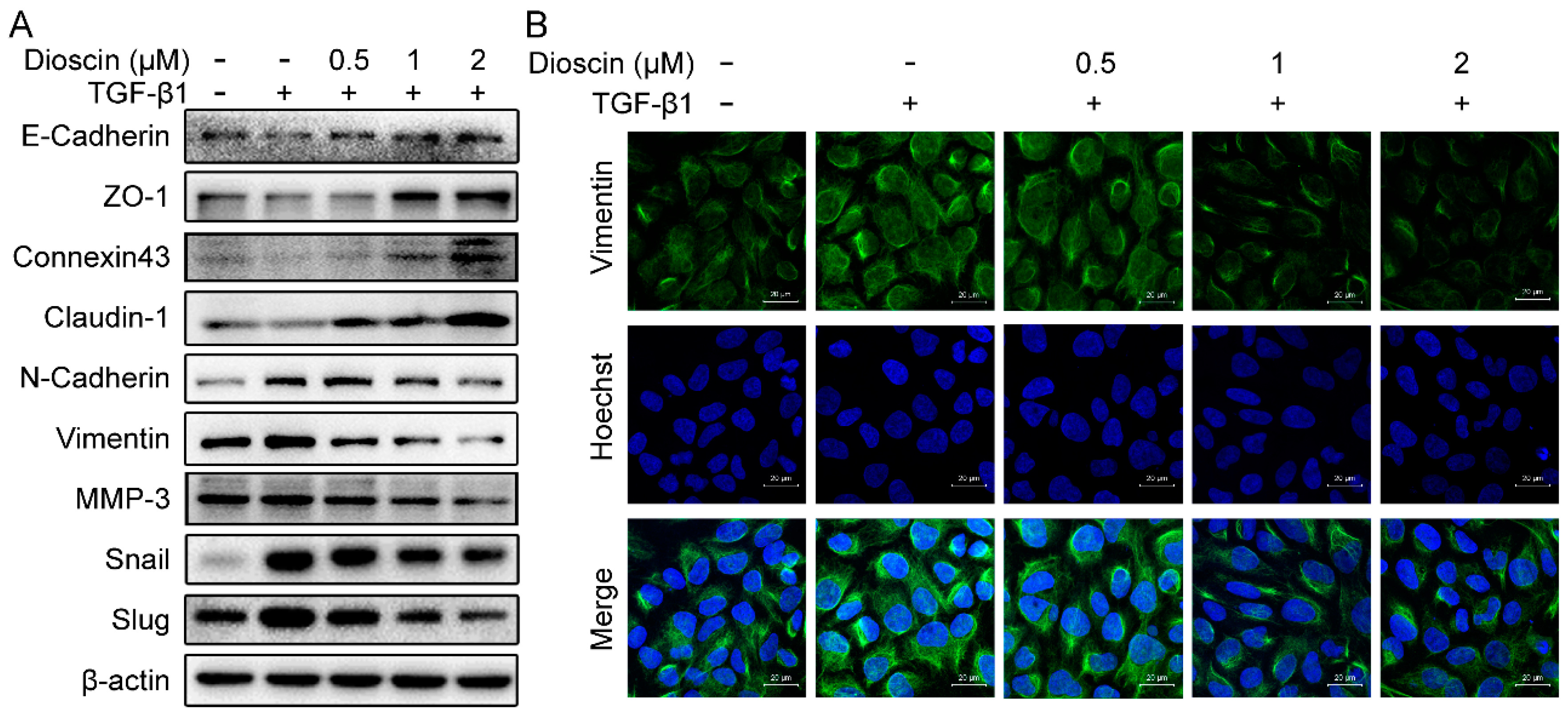
2.3. Effects of Dioscin on Expression of EMT Markers
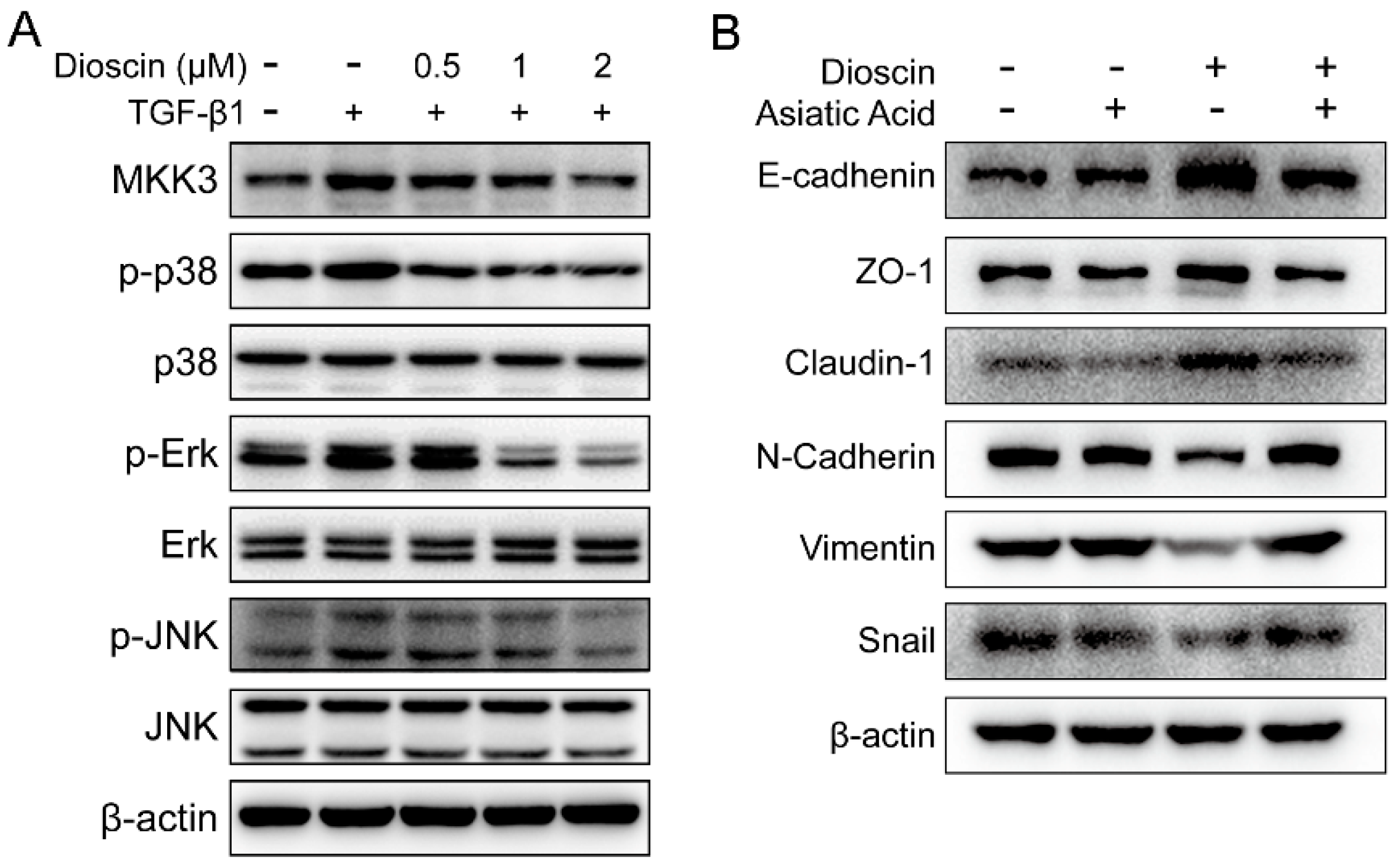
2.4. Dioscin Inhibits the MAPK Pathway.
3. Discussion
4. Materials and Methods
4.1. Drugs and Reagents
4.2. Cell Culture and Drug Treatments
4.3. Cell Viability Assay
4.4. Colony Formation Assay
4.5. Cell Cycle Analysis
4.6. CFDA SE Cell Proliferation Assay
4.7. 5-Ethynyl-20-deoxyuridine (EdU) Incorporation Assay
4.8. Western Blot Analysis
4.9. Wound Healing and Transwell Assays
4.10. Immunofluorescence Assay
4.11. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Xu, X.H.; Li, T.; Fong, C.M.; Chen, X.; Chen, X.J.; Wang, Y.T.; Huang, M.Q.; Lu, J.J. Saponins from Chinese Medicines as Anticancer Agents. Molecules 2016, 21, 1326. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tang, Y.M.; Yu, S.L.; Han, Y.W.; Kou, J.P.; Liu, B.L.; Yu, B.Y. Advances in the pharmacological activities and mechanisms of diosgenin. Chin. J. Nat. Med. 2015, 13, 578–587. [Google Scholar] [CrossRef]
- Tao, X.; Yin, L.; Xu, L.; Peng, J. Dioscin: A diverse acting natural compound with therapeutic potential in metabolic diseases, cancer, inflammation and infections. Pharmacol. Res. 2018, 137, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Xu, L.; Yin, L.; Han, X.; Qi, Y.; Xu, Y.; Song, S.; Zhao, Y.; Peng, J. Dioscin induces prostate cancer cell apoptosis through activation of estrogen receptor-beta. Cell Death & Disease 2017, 8, e2989. [Google Scholar]
- Song, X.; Wang, Z.; Liang, H.; Zhang, W.; Ye, Y.; Li, H.; Hu, Y.; Zhang, Y.; Weng, H.; Lu, J.; et al. Dioscin Induces Gallbladder Cancer Apoptosis by Inhibiting ROS-Mediated PI3K/AKT Signalling. Int. J. Biol. Sci. 2017, 13, 782–793. [Google Scholar] [CrossRef] [Green Version]
- Kou, Y.; Ji, L.; Wang, H.; Wang, W.; Zheng, H.; Zou, J.; Liu, L.; Qi, X.; Liu, Z.; Du, B.; et al. Connexin 43 upregulation by dioscin inhibits melanoma progression via suppressing malignancy and inducing M1 polarization. Int. J. Cancer 2017, 141, 1690–1703. [Google Scholar] [CrossRef] [Green Version]
- Bruix, J.; Gores, G.J.; Mazzaferro, V. Hepatocellular carcinoma: Clinical frontiers and perspectives. Gut 2014, 63, 844–855. [Google Scholar] [CrossRef]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Invest. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [Green Version]
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 2005, 6, 56. [Google Scholar] [CrossRef]
- Mulholland, D.J.; Kobayashi, N.; Ruscetti, M.; Zhi, A.; Tran, L.M.; Huang, J.; Gleave, M.; Wu, H. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 2012, 72, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.C.; Kim, H.; Kim, Y.J.; Choi, K.C.; Lee, I.H.; Lee, K.H.; Kim, M.K.; Ko, H. Dioscin suppresses TGF-beta1-induced epithelial-mesenchymal transition and suppresses A549 lung cancer migration and invasion. Bioorg. Med. Chem. Lett. 2017, 27, 3342–3348. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K.; Ehata, S.; Koinuma, D. Tumor-promoting functions of transforming growth factor-beta in progression of cancer. Upsala J. Med. Sci. 2012, 117, 143–152. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, A.K.; Jin, Y.; Luo, H.; Tang, M.; Pampo, C.; Shao, R.; Siemann, D.W.; Wu, L.; Heldermon, C.D.; Law, B.K.; et al. Epithelial-to-mesenchymal transition confers pericyte properties on cancer cells. J. Clin. Invest. 2016, 126, 4174–4186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Zhong, X.Y.; Zhang, L.H.; Jia, S.Q.; Shi, T.; Niu, Z.J.; Du, H.; Zhang, G.G.; Hu, Y.; Lu, A.P.; Li, J.Y.; et al. Positive association of up-regulated Cripto-1 and down-regulated E-cadherin with tumour progression and poor prognosis in gastric cancer. Histopathology 2008, 52, 560–568. [Google Scholar] [CrossRef]
- Toiyama, Y.; Yasuda, H.; Saigusa, S.; Tanaka, K.; Inoue, Y.; Goel, A.; Kusunoki, M. Increased expression of Slug and Vimentin as novel predictive biomarkers for lymph node metastasis and poor prognosis in colorectal cancer. Carcinogenesis 2013, 34, 2548–2557. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.; Robinson, M.; Smith, E.; Huntley, S.; Prime, S.; Paterson, I. Induction of an epithelial to mesenchymal transition in human immortal and malignant keratinocytes by TGF-beta1 involves MAPK, Smad and AP-1 signalling pathways. J. Cell. Biochem. 2005, 95, 918–931. [Google Scholar] [CrossRef]
- Guo, Y.; Jiang, M.; Zhao, X.; Gu, M.; Wang, Z.; Xu, S.; Yue, W. Cyclophilin A promotes non-small cell lung cancer metastasis via p38 MAPK. Thoracic cancer 2018, 9, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Steeg, P.S. Metastasis suppressors: Functional pathways. Lab. Invest. 2018, 98, 198–210. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the Dioscin are available from the authors. |




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, B.; Zhou, S.; Zhan, Y.; Ke, J.; Wang, K.; Liang, Q.; Hou, Y.; Zhu, P.; Ao, W.; Wei, X.; et al. Dioscin Inhibits the Invasion and Migration of Hepatocellular Carcinoma HepG2 Cells by Reversing TGF-β1-Induced Epithelial-Mesenchymal Transition. Molecules 2019, 24, 2222. https://doi.org/10.3390/molecules24122222
Chen B, Zhou S, Zhan Y, Ke J, Wang K, Liang Q, Hou Y, Zhu P, Ao W, Wei X, et al. Dioscin Inhibits the Invasion and Migration of Hepatocellular Carcinoma HepG2 Cells by Reversing TGF-β1-Induced Epithelial-Mesenchymal Transition. Molecules. 2019; 24(12):2222. https://doi.org/10.3390/molecules24122222
Chicago/Turabian StyleChen, Bonan, Shikun Zhou, Yujuan Zhan, Junzi Ke, Kun Wang, Qiqi Liang, Yu Hou, Pingping Zhu, Weizhen Ao, Xianli Wei, and et al. 2019. "Dioscin Inhibits the Invasion and Migration of Hepatocellular Carcinoma HepG2 Cells by Reversing TGF-β1-Induced Epithelial-Mesenchymal Transition" Molecules 24, no. 12: 2222. https://doi.org/10.3390/molecules24122222