Chenodeoxycholic Acid Ameliorates AlCl3-Induced Alzheimer’s Disease Neurotoxicity and Cognitive Deterioration via Enhanced Insulin Signaling in Rats

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
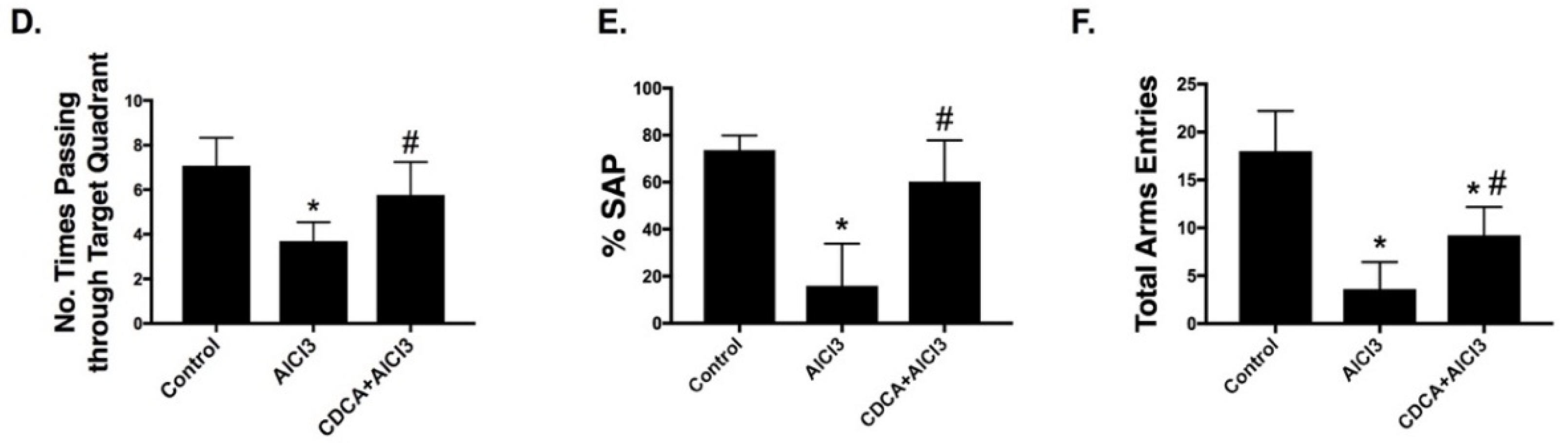
2.1. CDCA Improves Learning, Spatial Working Memory and General Activity in AD Rat Model
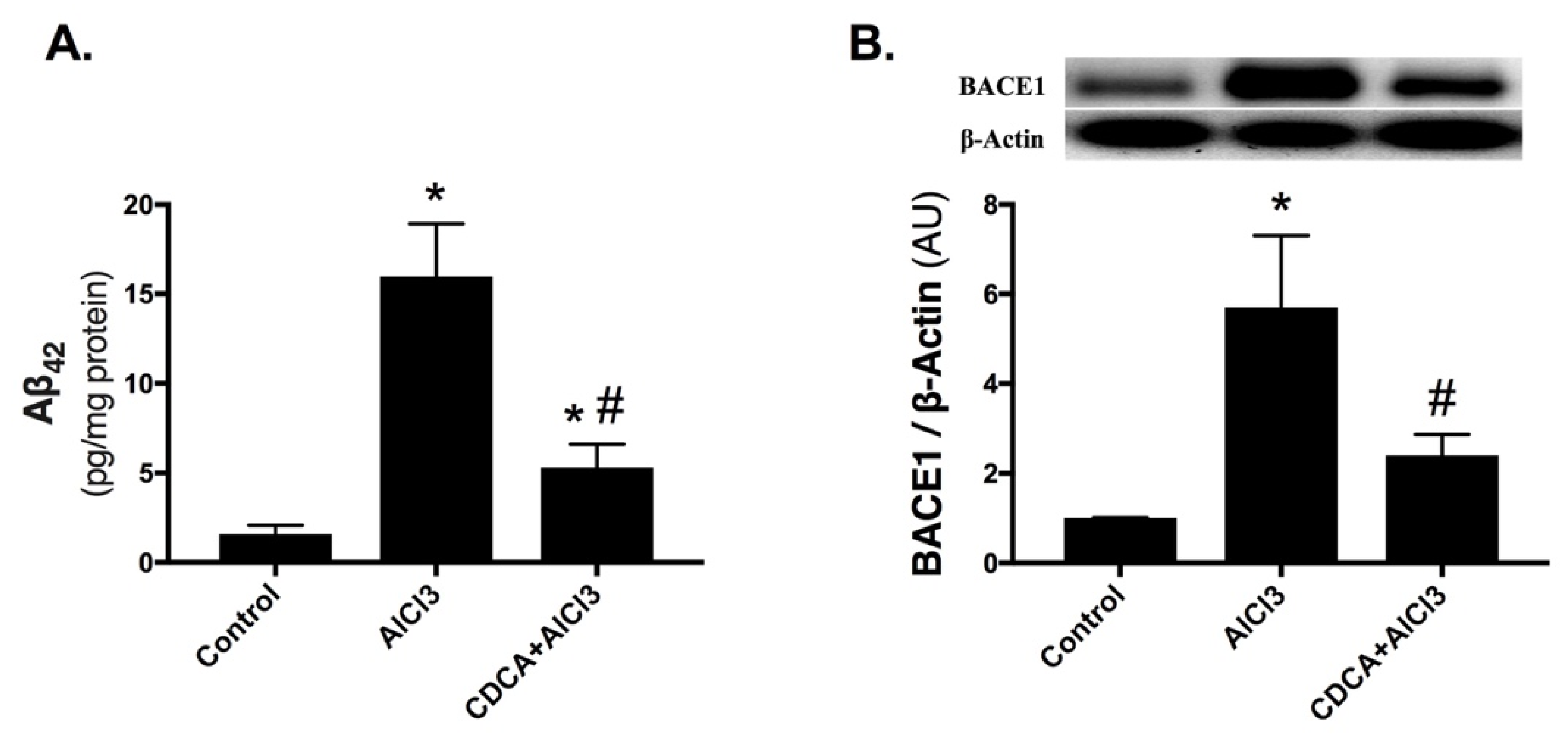
2.2. CDCA Decreases Amyloid-Beta Production in AD Rat Model
2.3. CDCA Augments Hippocampal Insulin Signaling in AD Rat Model
2.4. CDCA Improves Hippocampal GLP-1 and PPARγ Levels in AD Rat Model
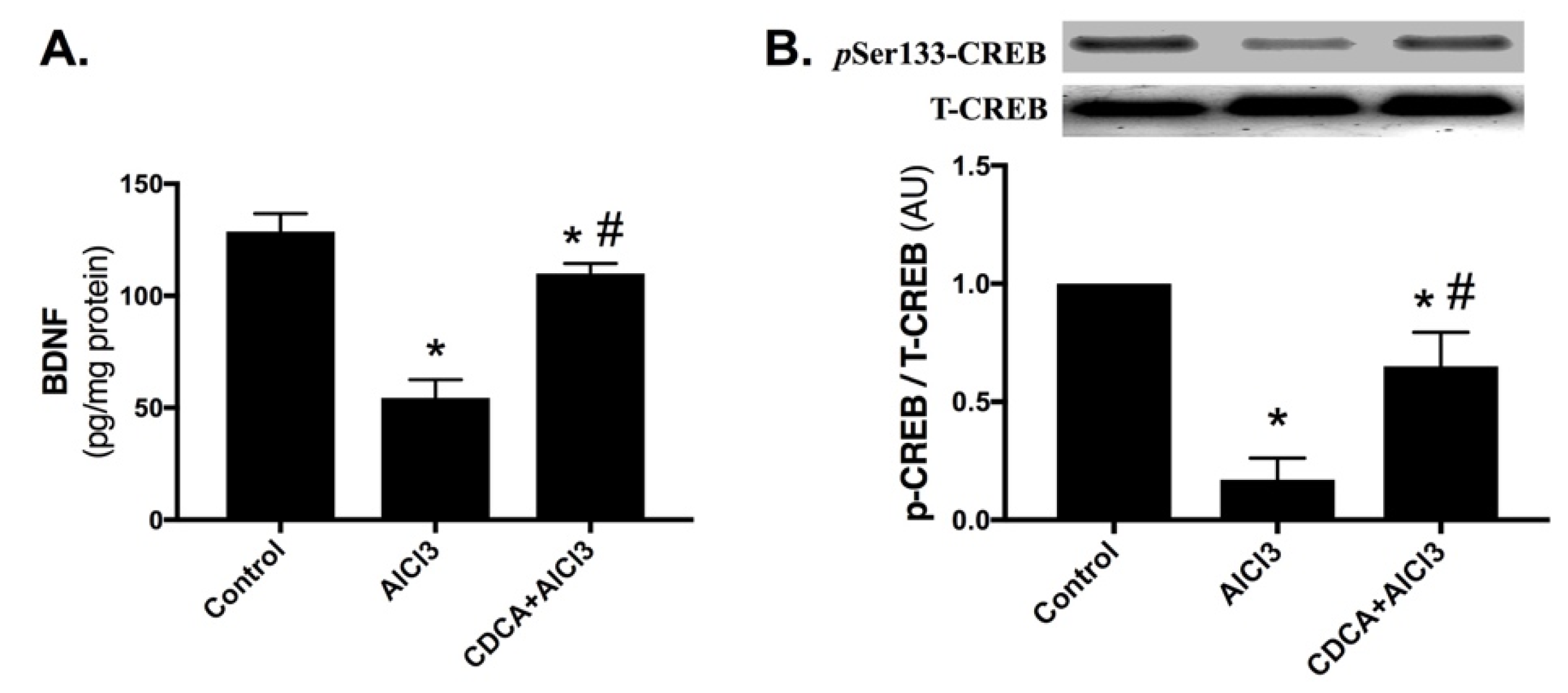
2.5. CDCA Promotes Hippocampal BDNF/CREB Pathway in AD Rat Model
2.6. CDCA Reduces AlCl3-Induced Neurodegeneration
3. Discussion
4. Materials and Methods
4.1. Animals and Ethical Considerations
4.2. Chemicals, Antibodies and Kits
4.3. Induction of Alzheimer’s Model and CDCA Dosing
4.4. Experimental Design
4.5. Behavioral Testing
4.5.1. Morris Water Maze
4.5.2. Y-Maze: Spontaneous Alternation
4.6. Tissue Sampling
4.7. Biochemical Analysis
4.8. Histopathological Examination
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. Alzheimers Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
- Bazzari, F.H.; Abdallah, D.M.; El-Abhar, H.S. Pharmacological Interventions to Attenuate Alzheimer’s Disease Progression: The Story So Far. Curr. Alzheimer Res. 2019, 16, 261–277. [Google Scholar] [CrossRef]
- Benedict, C.; Grillo, C.A. Insulin Resistance as a Therapeutic Target in the Treatment of Alzheimer’s Disease: A State-of-the-Art Review. Front. Neurosci. 2018, 12, 215. [Google Scholar] [CrossRef] [PubMed]
- Bedse, G.; Di Domenico, F.; Serviddio, G.; Cassano, T. Aberrant insulin signaling in Alzheimer’s disease: Current knowledge. Front. Neurosci. 2015, 9, 204. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Feldman, E.L. Insulin resistance as a key link for the increased risk of cognitive impairment in the metabolic syndrome. Exp. Mol. Med. 2015, 47, e149. [Google Scholar] [CrossRef]
- Forman, B.M.; Goode, E.; Chen, J.; Oro, A.E.; Bradley, D.J.; Perlmann, T.; Noonan, D.J.; Burka, L.T.; McMorris, T.; Lamph, W.W.; et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995, 81, 687–693. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lee, F.Y.; Barrera, G.; Lee, H.; Vales, C.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 1006–1011. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.; Saha, P.K.; Chan, L.; Moore, D.D. Farnesoid X receptor is essential for normal glucose homeostasis. J. Clin. Investig. 2006, 116, 1102–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cipriani, S.; Mencarelli, A.; Palladino, G.; Fiorucci, S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J. Lipid Res. 2010, 51, 771–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Huang, Y.; Yan, L.; Gao, M.; Liu, D. Synthetic FXR agonist GW4064 prevents diet-induced hepatic steatosis and insulin resistance. Pharm. Res. 2013, 30, 1447–1457. [Google Scholar] [CrossRef]
- Fang, S.; Suh, J.M.; Reilly, S.M.; Yu, E.; Osborn, O.; Lackey, D.; Yoshihara, E.; Perino, A.; Jacinto, S.; Lukasheva, Y.; et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat. Med. 2015, 21, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wang, J.; Hu, W.; Wang, C.; Lu, X.; Tong, L.; Wu, F.; Zhang, W. Identification of functional farnesoid X receptors in brain neurons. FEBS Lett. 2016, 590, 3233–3242. [Google Scholar] [CrossRef] [PubMed]
- Lew, J.L.; Zhao, A.; Yu, J.; Huang, L.; de Pedro, N.; Peláez, F.; Wright, S.D.; Cui, J. The farnesoid X receptor controls gene expression in a ligand-and promoter-selective fashion. J. Biol. Chem. 2004, 279, 8856–8861. [Google Scholar] [CrossRef]
- Quinn, M.; DeMorrow, S. Bile in the brain? A role for bile acids in the central nervous system. J. Cell Sci. 2012, 3, 7. [Google Scholar] [CrossRef]
- Frommherz, L.; Bub, A.; Hummel, E.; Rist, M.J.; Roth, A.; Watzl, B.; Kulling, S.E. Age-related changes of plasma bile acid concentrations in healthy adults—Results from the cross-sectional KarMeN study. PLoS ONE 2016, 11, e0153959. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudiandehkordi, S.; Arnold, M.; Nho, K.; Ahmad, S.; Jia, W.; Xie, G.; Louie, G.; Kueider-Paisley, A.; Moseley, M.A.; Thompson, J.W.; et al. Altered Bile Acid Profile Associates with Cognitive Impairment in Alzheimer’s Disease—An Emerging Role for Gut Microbiome. Alzheimers Dement. 2019, 15, 76–92. [Google Scholar] [CrossRef]
- Ackerman, H.D.; Gerhard, G.S. Bile acids in neurodegenerative disorders. Front. Aging Neurosci. 2016, 8, 263. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, R.M.; Nunes, A.F.; Dias, R.B.; Amaral, J.D.; Lo, A.C.; D’Hooge, R.; Sebastião, A.M.; Rodrigues, C.M. Tauroursodeoxycholic acid suppresses amyloid β-induced synaptic toxicity in vitro and in APP/PS1 mice. Neurobiol. Aging 2013, 34, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Salen, G.; Steiner, R.D. Epidemiology, diagnosis, and treatment of cerebrotendinous xanthomatosis (CTX). J. Inherit. Metab. Dis. 2017, 40, 771–881. [Google Scholar] [CrossRef]
- Alawdi, S.H.; El-Denshary, E.S.; Safar, M.M.; Eidi, H.; David, M.O.; Abdel-Wahhab, M.A. Neuroprotective effect of nanodiamond in Alzheimer’s disease rat model: A pivotal role for modulating NF-κB and STAT3 signaling. Mol. Neurobiol. 2017, 54, 1906–1918. [Google Scholar] [CrossRef]
- Giunta, S.; Andriolo, V.; Castorina, A. Dual blockade of the A1 and A2A adenosine receptor prevents amyloid beta toxicity in neuroblastoma cells exposed to aluminum chloride. Int. J. Biochem. Cell Biol. 2014, 54, 122–136. [Google Scholar] [CrossRef]
- Nampoothiri, M.; Kumar, N.; Ramalingayya, G.V.; Kutty, N.G.; Krishnadas, N.; Rao, C.M. Effect of insulin on spatial memory in aluminum chloride-induced dementia in rats. Neuroreport 2017, 28, 540–544. [Google Scholar] [CrossRef]
- Frölich, L.; Blum-Degen, D.; Bernstein, H.G.; Engelsberger, S.; Humrich, J.; Laufer, S.; Muschner, D.; Thalheimer, A.; Türk, A.; Hoyer, S.; et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J. Neural Transm. 1998, 105, 423–438. [Google Scholar] [CrossRef]
- Muller, A.P.; Gnoatto, J.; Moreira, J.D.; Zimmer, E.R.; Haas, C.B.; Lulhier, F.; Perry, M.L.; Souza, D.O.; Torres-Aleman, I.; Portela, L.V. Exercise increases insulin signaling in the hippocampus: Physiological effects and pharmacological impact of intracerebroventricular insulin administration in mice. Hippocampus 2011, 21, 1082–1092. [Google Scholar] [CrossRef]
- Justin-Thenmozhi, A.; Dhivya Bharathi, M.; Kiruthika, R.; Manivasagam, T.; Borah, A.; Essa, M.M. Attenuation of Aluminum Chloride-Induced Neuroinflammation and Caspase Activation Through the AKT/GSK-3β Pathway by Hesperidin in Wistar Rats. Neurotox. Res. 2018, 34, 463–476. [Google Scholar] [CrossRef]
- Wei, X.; Wei, H.; Yang, D.; Li, D.; Yang, X.; He, M.; Lin, E.; Wu, B. Effect of Aluminum Exposure on Glucose Metabolism and Its Mechanism in Rats. Biol. Trace Elem. Res. 2018, 186, 450–456. [Google Scholar] [CrossRef]
- Teodoro, J.S.; Rolo, A.P.; Jarak, I.; Palmeira, C.M.; Carvalho, R.A. The bile acid chenodeoxycholic acid directly modulates metabolic pathways in white adipose tissue in vitro: Insight into how bile acids decrease obesity. NMR Biomed. 2016, 29, 1391–1402. [Google Scholar] [CrossRef]
- Shihabudeen, M.S.; Roy, D.; James, J.; Thirumurugan, K. Chenodeoxycholic acid, an endogenous FXR ligand alters adipokines and reverses insulin resistance. Mol. Cell. Endocrinol. 2015, 414, 19–28. [Google Scholar] [CrossRef]
- Zhang, H.M.; Wang, X.; Wu, Z.H.; Liu, H.L.; Chen, W.; Zhang, Z.Z.; Chen, D.; Zeng, T.S. Beneficial effect of farnesoid X receptor activation on metabolism in a diabetic rat model. Mol. Med. Rep. 2016, 13, 2135–2142. [Google Scholar] [CrossRef]
- Shen, H.; Zhang, Y.; Ding, H.; Wang, X.; Chen, L.; Jiang, H.; Shen, X. Farnesoid X receptor induces GLUT4 expression through FXR response element in the GLUT4 promoter. Cell. Physiol. Biochem. 2008, 22, 1–14. [Google Scholar] [CrossRef]
- Renga, B.; Mencarelli, A.; Vavassori, P.; Brancaleone, V.; Fiorucci, S. The bile acid sensor FXR regulates insulin transcription and secretion. Biochim. Biophys. Acta 2010, 1802, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 574–582. [Google Scholar] [CrossRef]
- Zhao, W.Q.; De Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008, 22, 246–260. [Google Scholar] [CrossRef]
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative toxicity in diabetes and Alzheimer’s disease: Mechanisms behind ROS/RNS generation. J. Biomed. Sci. 2017, 24, 76. [Google Scholar] [CrossRef]
- Clark, I.; Atwood, C.; Bowen, R.; Paz-Filho, G.; Vissel, B. Tumor necrosis factor-induced cerebral insulin resistance in Alzheimer’s disease links numerous treatment rationales. Pharm. Rev. 2012, 64, 1004–1026. [Google Scholar] [CrossRef]
- Aguirre, V.; Werner, E.D.; Giraud, J.; Lee, Y.H.; Shoelson, S.E.; White, M.F. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J. Biol. Chem. 2002, 277, 1531–1537. [Google Scholar] [CrossRef]
- Yarchoan, M.; Toledo, J.B.; Lee, E.B.; Arvanitakis, Z.; Kazi, H.; Han, L.Y.; Louneva, N.; Lee, V.M.Y.; Kim, S.F.; Trojanowski, J.Q.; et al. Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer’s disease and tauopathies. Acta Neuropathol. 2014, 128, 679–689. [Google Scholar] [CrossRef]
- Kaminari, A.; Giannakas, N.; Tzinia, A.; Tsilibary, E.C. Overexpression of matrix metalloproteinase-9 (MMP-9) rescues insulin-mediated impairment in the 5XFAD model of Alzheimer’s disease. Sci. Rep. 2017, 7, 683. [Google Scholar] [CrossRef]
- Nielsen, S.; Svane, M.S.; Kuhre, R.E.; Clausen, T.R.; Kristiansen, V.B.; Rehfeld, J.F.; Holst, J.J.; Madsbad, S.; Bojsen-Moller, K.N. Chenodeoxycholic acid stimulates glucagon-like peptide-1 secretion in patients after Roux-en-Y gastric bypass. Physiol. Rep. 2017, 5, e13140. [Google Scholar] [CrossRef]
- Katsurada, K.; Yada, T. Neural effects of gut-and brain-derived glucagon-like peptide-1 and its receptor agonist. J. Diabetes Investig. 2016, 7, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Brighton, C.A.; Rievaj, J.; Kuhre, R.E.; Glass, L.L.; Schoonjans, K.; Holst, J.J.; Gribble, F.M.; Reimann, F. Bile acids trigger GLP-1 release predominantly by accessing basolaterally located G protein–coupled bile acid receptors. Endocrinology 2015, 156, 3961–3970. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Scheltema, M.J.; Sonne, D.P.; Hansen, J.S.; Sperling, M.; Rehfeld, J.F.; Holst, J.J.; Vilsbøll, T.; Knop, F.K. Effect of chenodeoxycholic acid and the bile acid sequestrant colesevelam on glucagon-like peptide-1 secretion. Diabetes Obes. Metab. 2016, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Macchiarulo, A.; Thomas, C.; Gioiello, A.; Une, M.; Hofmann, A.F.; Saladin, R.; Schoonjans, K.; Pellicciari, R.; Auwerx, J. Novel potent and selective bile acid derivatives as TGR5 agonists: Biological screening, structure-activity relationships, and molecular modeling studies. J. Med. Chem. 2008, 51, 1831–1841. [Google Scholar] [CrossRef]
- Li, S.; Qiu, M.; Kong, Y.; Zhao, X.; Choi, H.J.; Reich, M.; Bunkelman, B.H.; Liu, Q.; Hu, S.; Han, M.; et al. Bile Acid G Protein-Coupled Membrane Receptor TGR5 Modulates Aquaporin 2–Mediated Water Homeostasis. J. Am. Soc. Nephrol. 2018, 29, 2658–2670. [Google Scholar] [CrossRef]
- Pathak, P.; Liu, H.; Boehme, S.; Xie, C.; Krausz, K.W.; Gonzalez, F.; Chiang, J.Y. Farnesoid X receptor induces Takeda G-protein receptor 5 crosstalk to regulate bile acid synthesis and hepatic metabolism. J. Biol. Chem. 2017, 292, 11055–11069. [Google Scholar] [CrossRef]
- Malinowski, J.M.; Bolesta, S. Rosiglitazone in the treatment of type 2 diabetes mellitus: A critical review. Clin. Ther. 2000, 22, 1151–1168. [Google Scholar] [CrossRef]
- Zheng, F.; Cai, Y. Concurrent exercise improves insulin resistance and nonalcoholic fatty liver disease by upregulating PPAR-γ and genes involved in the beta-oxidation of fatty acids in ApoE-KO mice fed a high-fat diet. Lipids Health Dis. 2019, 18, 6. [Google Scholar] [CrossRef]
- Fiorucci, S.; Rizzo, G.; Antonelli, E.; Renga, B.; Mencarelli, A.; Riccardi, L.; Morelli, A.; Pruzanski, M.; Pellicciari, R. Cross-talk between farnesoid-X-receptor (FXR) and peroxisome proliferator-activated receptor γ contributes to the antifibrotic activity of FXR ligands in rodent models of liver cirrhosis. J. Pharm. Exp. 2005, 315, 58–68. [Google Scholar] [CrossRef]
- Xin, X.M.; Zhong, M.X.; Yang, G.L.; Peng, Y.; Zhang, Y.L.; Zhu, W. GW4064, a farnesoid X receptor agonist, upregulates adipokine expression in preadipocytes and HepG2 cells. World J. Gastroenterol. 2014, 20, 15727–15735. [Google Scholar] [CrossRef]
- Borba, E.M.; Duarte, J.A.; Bristot, G.; Scotton, E.; Camozzato, A.L.; Chaves, M.L. Brain-derived neurotrophic factor serum levels and hippocampal volume in mild cognitive impairment and dementia due to Alzheimer disease. Dement. Geriatr. Cogn. Dis. Extra 2016, 6, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Zhuge, W.; Yang, J.; Wen, F.; Xu, Z.; Wang, X.; Zhuge, Q. Insulin resistance disrupts the interaction between AKT and the NMDA receptor and the inactivation of the CaMKIV/CREB pathway in minimal hepatic encephalopathy. Toxicol. Sci. 2017, 159, 290–306. [Google Scholar] [CrossRef] [PubMed]
- Suwa, M.; Yamamoto, K.I.; Nakano, H.; Sasaki, H.; Radak, Z.; Kumagai, S. Brain-derived neurotrophic factor treatment increases the skeletal muscle glucose transporter 4 protein expression in mice. Physiol. Res. 2010, 59, 619–623. [Google Scholar]
- Bathina, S.; Das, U.N. Brain-derived neurotrophic factor and its clinical implications. Arch. Med. Sci. 2015, 11, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Kariharan, T.; Nanayakkara, G.; Parameshwaran, K.; Bagasrawala, I.; Ahuja, M.; Abdel-Rahman, E.; Amin, A.T.; Dhanasekaran, M.; Suppiramaniam, V.; Amin, R.H. Central activation of PPAR-gamma ameliorates diabetes induced cognitive dysfunction and improves BDNF expression. Neurobiol. Aging 2015, 36, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.; Zhang, J.; Li, C.Y.; Wang, Y.; Zeng, M.J.; Cai, Z.X.; Tian, R.B.; Jia, W.; Li, X.H. Insulin resistance-induced hyperglycemia decreased the activation of Akt/CREB in hippocampus neurons: Molecular evidence for mechanism of diabetes-induced cognitive dysfunction. Neuropeptides 2015, 54, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Autry, A.E.; Monteggia, L.M. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharm. Rev. 2012, 64, 238–258. [Google Scholar] [CrossRef] [PubMed]
- National Research Council. Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Walton, J.R.; Wang, M.X. APP expression, distribution and accumulation are altered by aluminum in a rodent model for Alzheimer’s disease. J. Inorg. Biochem. 2009, 103, 1548–1554. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br. J. Pharm. 2010, 160, 1577–1579. [Google Scholar] [CrossRef]
- Duell, P.B.; Salen, G.; Eichler, F.S.; DeBarber, A.E.; Connor, S.L.; Casaday, L.; Jayadev, S.; Kisanuki, Y.; Lekprasert, P.; Malloy, M.J.; et al. Diagnosis, Treatment and Clinical Outcomes in 43 Cases with Cerebrotendinous Xanthomatosis. J. Clin. Lipidol. 2018, 12, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [Green Version]
- LEON, M.P.; Loria, P.; Carulli, N.; Murphy, G.M.; DOWLING, R.H. Intestinal solubilization, absorption, pharmacokinetics and bioavailability of chenodeoxycholic acid. Eur. J. Clin. Investig. 1980, 10, 261–271. [Google Scholar] [CrossRef]
- Cavas, M.; Beltrán, D.; Navarro, J.F. Behavioural effects of dimethyl sulfoxide (DMSO): Changes in sleep architecture in rats. Toxicol. Lett. 2005, 157, 221–232. [Google Scholar] [CrossRef]
- Pascual, M.; Blanco, A.M.; Cauli, O.; Miñarro, J.; Guerri, C. Intermittent ethanol exposure induces inflammatory brain damage and causes long-term behavioural alterations in adolescent rats. Eur. J. Neurosci. 2007, 25, 541–550. [Google Scholar] [CrossRef]
- European Medicines Agency. Chenodeoxycholic Acid Leadiant. ANNEX I: Summary of Product Characteristics. Available online: https://www.ema.europa.eu/documents/product-information/chenodeoxycholic-acid-leadiant-epar-product-information_en.pdf (accessed on 25 March 2019).
- Moshtaghie, A.A.; Malekpouri, P.; Moshtaghie, M.; Mohammadi-nejad, M.; Ani, M. Protective effects of copper against aluminum toxicity on acetylcholinesterase and catecholamine contents of different regions of rat’s brain. Neurol. Sci. 2013, 34, 1639–1650. [Google Scholar] [CrossRef]
- Al-Otaibi, S.S.; Arafah, M.M.; Sharma, B.; Alhomida, A.S.; Siddiqi, N.J. Synergistic Effect of Quercetin and α-Lipoic Acid on Aluminium Chloride Induced Neurotoxicity in Rats. J. Toxicol. 2018, 2018, 2817036. [Google Scholar] [CrossRef]
- Çolak, S.; Geyikoğlu, F.; Keles, O.N.; Türkez, H.; Topal, A.; Unal, B. The neuroprotective role of boric acid on aluminum chloride-induced neurotoxicity. Toxicol. Ind. Health 2011, 27, 700–710. [Google Scholar] [CrossRef]
- Baydar, T.; Papp, A.; Aydin, A.; Nagymajtenyi, L.; Schulz, H.; Isimer, A.; Sahin, G. Accumulation of aluminum in rat brain. Biol. Trace Elem. Res. 2003, 92, 231–244. [Google Scholar] [CrossRef]
- Morris, R. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 1984, 11, 47–60. [Google Scholar] [CrossRef]
- Vorhees, C.V.; Williams, M.T. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef]
- Ghafouri, S.; Fathollahi, Y.; Javan, M.; Shojaei, A.; Asgari, A.; Mirnajafi-Zadeh, J. Effect of low frequency stimulation on impaired spontaneous alternation behavior of kindled rats in Y-maze test. Epilepsy Res. 2016, 126, 37–44. [Google Scholar] [CrossRef]
- Stanford Medicine. Y Maze Spontaneous Alternation Test. Available online: https://med.stanford.edu/sbfnl/services/bm/lm/y-maze.html (accessed on 28 March 2019).
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Bancroft, J.D.; Layton, C. The Hematoxylins and Eosin. In Bancroft’s Theory and Practice of Histological Techniques, 8th ed.; Suvarna, S.K., Layton, C., Bancroft, J.D., Eds.; Elsevier Health Sciences: London, UK, 2018; pp. 126–138. [Google Scholar]
Sample Availability: Samples of the compounds are not available. |









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bazzari, F.H.; Abdallah, D.M.; El-Abhar, H.S. Chenodeoxycholic Acid Ameliorates AlCl3-Induced Alzheimer’s Disease Neurotoxicity and Cognitive Deterioration via Enhanced Insulin Signaling in Rats. Molecules 2019, 24, 1992. https://doi.org/10.3390/molecules24101992
Bazzari FH, Abdallah DM, El-Abhar HS. Chenodeoxycholic Acid Ameliorates AlCl3-Induced Alzheimer’s Disease Neurotoxicity and Cognitive Deterioration via Enhanced Insulin Signaling in Rats. Molecules. 2019; 24(10):1992. https://doi.org/10.3390/molecules24101992
Chicago/Turabian StyleBazzari, Firas H., Dalaal M. Abdallah, and Hanan S. El-Abhar. 2019. "Chenodeoxycholic Acid Ameliorates AlCl3-Induced Alzheimer’s Disease Neurotoxicity and Cognitive Deterioration via Enhanced Insulin Signaling in Rats" Molecules 24, no. 10: 1992. https://doi.org/10.3390/molecules24101992