Solid-Phase Synthesis of Phosphorothioate/Phosphonothioate and Phosphoramidate/Phosphonamidate Oligonucleotides

Institute of Organic Chemistry and Biochemistry AS CR, v.v.i., Flemingovo nám. 2, 166 10 Prague 6, Czech Republic
*
Authors to whom correspondence should be addressed.
Molecules 2019, 24(10), 1872; https://doi.org/10.3390/molecules24101872
Submission received: 1 May 2019
/
Revised: 9 May 2019
/
Accepted: 14 May 2019
/
Published: 15 May 2019
(This article belongs to the Special Issue Antisense Oligonucleotide Chemistry and Applications)
Abstract
:We have developed a robust solid-phase protocol which allowed the synthesis of chimeric oligonucleotides modified with phosphodiester and O-methylphosphonate linkages as well as their P-S and P-N variants. The novel O-methylphosphonate-derived modifications were obtained by oxidation, sulfurization, and amidation of the O-methyl-(H)-phosphinate internucleotide linkage introduced into the oligonucleotide chain by H-phosphonate chemistry using nucleoside-O-methyl-(H)-phosphinates as monomers. The H-phosphonate coupling followed by oxidation after each cycle enabled us to successfully combine H-phosphonate and phosphoramidite chemistries to synthesize diversely modified oligonucleotide strands.
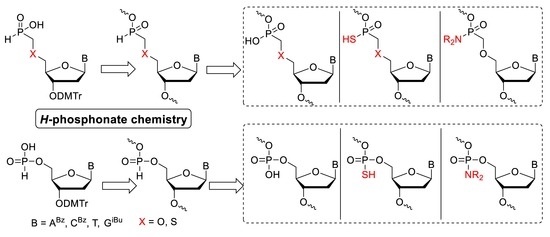
1. Introduction
Recently, we published a detailed study on the influence of the incorporation of 2′-deoxy-nucleoside 3′-O- and 5′-O-methylphosphonate units on the hybridization properties of modified DNA strands and on their ability to elicit E. coli RNase H activity in heteroduplexes [1]. Although the insertion of the bridging –CH2– group into the phosphodiester linkage should increase the total entropy of the system due to an additional degree of freedom, leading to duplex destabilization, only the incorporation of 3′-O-methylphosphonate units into the DNA strand decreased the stability of the appropriate heteroduplexes. In contrast, the presence of 5′-O-methylphosphonate units slightly stabilized the modifDNA*RNA heteroduplexes. These oligonucleotides, known as MethylPhosphonate Nucleic Acids (MePNA), when they contained various ratios of nucleoside-5′-phosphate and 5′-O-methyl-phosphonate units in an alternating mode exhibited superior enhancement of the RNase H cleavage rate.
To improve the synthesis of MePNA, we developed the straightforward synthesis of nucleoside-O-methyl-(H)-phosphinates and 5′-deoxynucleoside-5′-S-methyl-(H)-phosphinates [2,3] that were closely related to the well-known nucleoside H-phosphonates, [4,5] and demonstrated their compatibility with the H-phosphonate chemistry of the oligonucleotide synthesis. Recently, Herdewijn [6] has exploited this methodology to introduce various nucleoside phosphonates into oligonucleotides.
In this study, we present the extension of our methodology on the synthesis of modified oligonucleotides by a combination of phosphoramidite and H-phosphonate chemistries using nucleoside phosphoramidite, and H-phosphonate 1, nucleoside-O-methyl-(H)-phosphinate, and 5′-deoxynucleoside-5′-S-methyl-(H)-phosphinate monomers 2 as building blocks (Figure 1), respectively. Standard phosphoramidite chemistry allows the synthesis of phosphate 3, and phosphorothioate 4 internucleotide linkages. H-phosphonate chemistry affords even wider possibilities since the H-phosphonate internucleotide linkage in 5 can be oxidized, sulfurized, or amidated to form appropriate phosphate 3 [7,8], phosphorothioate 4 [9] or phosphoramidate 6 [10] internucleotide linkages (Figure 1). To be able to synthesize oligonucleotides modified with phosphodiester, O-methylphosphonate, and 5′-deoxy-5′-S-methylphosphonate linkages, we developed procedures which allowed the synthesis of H-phosphinate internucleotide linkage 7 (Figure 1). This linkage could be oxidized, sulfurized, or amidated to form the O-methylphosphonate 8a, O-methylphosphonothioate 9a, O-methylphosphonamidate 10, S-methylphosphonate 8b and S-methylphosphonothioate 9b internucleotide linkages, respectively. The insertion of the oxidation step after each H-phosphonate coupling step [11,12] allowed us to combine easily phosphonate-derived units with the phosphodiester ones and to prepare unique set of modified oligonucleotides.
2. Results and Discussion
2.1. Study on Model Dimers
Our results on novel nucleoside-O-methyl-(H)-phosphinates [2,3] as monomers for H-phosphonate chemistry prompted us to develop robust synthetic protocols that would allow the synthesis of oligonucleotides bearing any combination of phosphodiester 3, phosphorothioate 4, phosphoramidate 6, O-methylphosphonate 8a, O-methylphosphonothioate 9a, O-methyl-phosphonamidate 10, S-methylphosphonate 8b and S-methylphosphonothioate 9b internucleotide linkages (Figure 1). The optimization of the oxidative couplings affording the abovementioned modified bonds was performed on a model dimer. Thus, TentaGel modified with 4-N-benzoyl-2′-deoxy-3′-O-DMTr-cytidine-5′-hemisuccinate 11 was extended with thymidine-O-methyl-(H)-phosphinate or thymidine-5′-deoxy-5′-S-methyl-(H)-phosphinate monomer 2 to provide TentaGel-attached (dCT) dimer with H-phosphinate internucleotide linkage 12b. This linkage was subjected to oxidative procedures (oxidation/sulfurization/amidation) under various conditions (Scheme 1). All experiments with H-phosphinate monomers 2 were always compared to the standard thymidine H-phosphonate 1 as a control of the efficacy of the reaction conditions (For more details see Supporting Information).
2.1.1. H-Phosphonate Coupling
The optimization of coupling reaction of H-phosphinate monomers 2 [2,3] was performed at 0.1 M concentration in acetonitrile-pyridine mixture (1:1) with a series of activating agents [13] commonly used in H-phosphonate chemistry such as pivaloyl chloride, adamantanecarbonyl chloride, 2-chloro-5,5-dimethyl-1,3,2-dioxaphosphorinane 2-oxide (DMOCP), diphenyl chloro-phosphate (DPCP), and bis(2-oxo-3-oxazolidinyl)phosphinic chloride (OXP) at 0.3 M concentration in acetonitrile-pyridine mixtures (95:5). Of all tested coupling agents, we selected DMOCP as the activator of choice for the condensation of both H-phosphonate 1 and H-phosphinates 2. The only difference was the duration of the coupling step. The H-phosphinate monomers required 10 min condensation time compared to 5 min H-phosphonate condensation.
2.1.2. Oxidation/Amidation/Sulfurization of H-Phosphinate Internucleotide Linkage
As we published previously [2,3], the use of water-containing oxidation mixtures such as 0.1 M iodine in pyridine-water mixture (98:2) reported for the oxidation of H-phosphonate linkage 12a [7,8] could not be used for the oxidation of H-phosphinate bond 12b due to its lability under aqueous conditions and very fast hydrolytic cleavage. The use of 0.1 M iodine in pyridine-methanol (50:50) mixture led to only 80% yield of the expected MeO-P(V) product. However, Atherton-Todd reaction [14,15,16] conditions (CCl4-pyridine-methanol) provided an acceptable 95% yield of MeO-P(V) product. Moreover, no hydrolysis of internucleotide linkage was observed.
Our further research focused on the composition of the CCl4-based oxidation mixture, especially with respect to the base used and its basicity/nucleophilicity. The replacement of pyridine with 1-methylimidazole or 1-methylimidazole-triethylamine mixture, led to a quantitative oxidation of both H-phosphonate and H-phosphinate linkages. We found that methanol and 3-hydroxypropionitrile exhibited similar reactivity to provide methyl and 2-cyanoethyl esters, respectively, however, the 2-cyanoethyl ester represented more stable ester group. Since 3-hydroxypropionitrile is immiscible with tetrachloromethane, DCM was used as a co-solvent in oxidation mixture (Table 1).
Encouraged by these results and based on the fact that the amines were commonly used nucleophiles in Atherton-Todd reaction [15,16], we examined the preparation of electroneutral oligonucleotides with non-bridging P–N bond using oxidative amidation of the H-phosphinate linkage. In contrast to the reported phosphoramidate oligonucleotides [14,17,18], the phosphonamidate oligonucleotides have not been published so far. Two representative amines, the primary N,N-dimethylethylenediamine which was already incorporated into phosphoramidate oligonucleotides [17,18] and morpholine [19] as a representative of secondary amines were selected as amidation reagent. We examined various ratios of tetrachloromethane and the amines in the mixture, and the time of the amidation reaction. A quantitative amidation of both H-phosphonate and H-phosphinate linkages was achieved after 180 min using a mixture of CCl4/amine/DCM in 3:2:5 ratio (DCM was used to keep forming amine hydrochloride in the solution).
The sulfurization of H-phosphinate 12b linkage to afford phosphonothioate bond was achieved using two methods. The first method employed elemental sulfur in various solvents and in the presence of base [9], and provided directly expected charged dimers. The reaction performed with 0.5 M solution of elemental sulfur in pyridine provided the desired products quantitatively within 20 min. Since this type of sulfurization could be only used as the last step of the synthetic cycles, we also focused on the second method employing sulfur-transfer reagent (N-[(2-cyanoethyl)-sulfanyl]succinimide, CSS) [20,21]. This method was applicable either in the last H-phosphonate cycle or after each synthetic cycle affording S-(2-cyanoethyl) esters which were then cleaved by β-elimination to provide phosphonothioate linkages. Thus the sulfurization performed with 0.2 M CSS in a mixture of acetonitrile and silylating agent BSTFA [20] (29:1) provided the S-(2-cyanoethyl)-protected dimers quantitatively within 30 min. β-Elimination reaction was performed at the end of the synthesis with 1 M DBU in acetonitrile for 3 min. The optimal reaction conditions of the individual oxidation steps are summarized in Table 1.
2.1.3. Stability of Modified Internucleotide Linkages
During the optimization of the condensation and oxidation steps, we observed different behavior and reactivity of both H-phosphonate and H-phosphinate internucleotide linkages. Therefore we decided to examine the stability of these linkages using a series of modified d(CT) dimers. All experiments were performed on LCMS at several time intervals.
The stability of phosphorothioate and phosphonothioate linkages was examined in the presence of two potential desulfurizing agents—sodium periodate (0.05 M aqueous solution) and iodine (0.05 M solution in pyridine-water mixture (98:2)).
It was reported that the periodate anion partially desulfurized the phosphorothioate TpT dimer [22]. In agreement with this, the treatment of phosphorothioate d(CT) dimer 14a with sodium periodate quantitatively afforded the product of desulfurization, d(CT) phosphate dimer 15a, after 12 h. In case of phosphonothioate dimer 14b, complete cleavage of the internucleotide linkage to phosphonate 16b was observed. On the other hand, S-methylthiophosphonate dimer 14c was cleaved to phosphonate 16c only partially (53%). The rest of the dimer underwent desulfurization and oxidation of S-methylphosphonate linkage to S(=O)-methylphosphonate 15d (47%).
In case of iodine-promoted desulfurization, all tested dimers 14a–c were quantitatively and cleanly transformed to phosphate/phosphonate dimers 15a–c within 5 min of treatment. Neither cleavage nor oxidation of S-methylphosphonate in the case of dimer 14c was observed (Scheme 2).
We also tested the stability of P–N bonds of dimers 17e–h under acidic conditions (pH = 1 for 5 h; Scheme 2). Comparing the stability of phosphoramidate and phosphonamidate linkages, phosphoromorpholidate dimer 17e exhibited higher stability (35% hydrolysis to dimer 15a) than phosphonomorpholidate dimer 17f (78% hydrolysis to dimer 15b). Suprisingly, S-methyl-phosphonomorpholidate dimer 17g showed the highest stability, with only 18% hydrolysis to dimer 15c.
Moreover, phosphonamidate linkages derived from primary amines were much more stable than the linkages derived from secondary amines (Scheme 2). Dimethylethylendiamino-phosphonamidate dimer 17i and dimethylethylendiamino-S-methylphosphonamidate dimer 17j exhibited similar stability, with only 6% hydrolysis to the dimers 15b and 15c, respectively. Dimethylethylendiamino-phosphoramidate dimer 17h was hydrolyzed to the same extent as its morpholino derivative 17e (36% hydrolysis to dimer 15a).
2.2. Synthetic Protocols for Oligonucleotide Synthesis
Based on the results obtained on the level of dimers, we have developed three synthetic protocols (Synthetic protocols A, B, and C), and tested their efficacy on model homo-oligothymidylates dT101–dT107 modified with various internucleotide linkages (Table 2).
2.2.1. Synthetic Protocol A
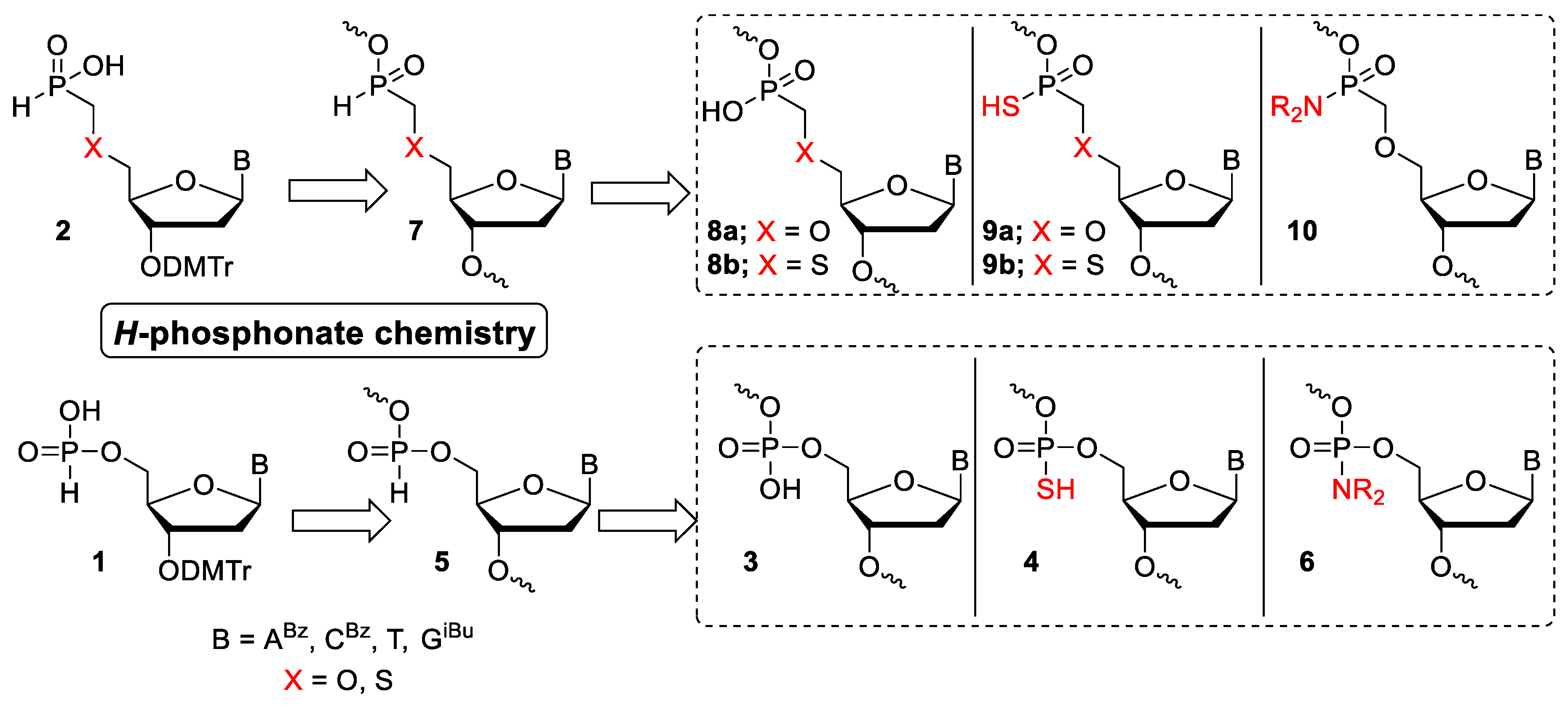
This protocol was used for the synthesis of oligonucleotides fully modified with either phosphonate/phosphate 18 or thiophosphonate/thiophosphate 19 linkages (Scheme 3, Table 2). The nucleoside H-phosphonates 1 and O-methyl-(H)-phosphinates 2 were coupled on solid support using H-phosphonate chemistry, to provide oligonucleotide 20. Since the use of H-phosphonate capping [23] led, in our hands, to the formation of substantial amount of side products, this step was omitted. After last synthetic cycle, all internucleotide linkages were either oxidized or sulfurized to afford oligonucleotides 21 or 19, respectively (Scheme 3). The use of non-aqueous oxidation mixtures (Table 1; Ox-A, Ox-B, and Ox-C) that oxidized P–H bonds to P–OR bond (R = CH3, 2-cyanoethyl) led to complete oxidation. The methyl and 2-cyanoethyl ester groups were easily removed using thiophenol/TEA/DMF and gaseous ammonia, respectively, to afford the final product 18.
The sulfurization of the H-phosphonate/H-phosphinate linkages proceeded smoothly using a solution of elemental sulfur in pyridine (Table 1; Ox-D). It is important to note that the elimination of traces of water from the oxidation and sulfurization mixtures was of the utmost importance. Therefore, all the oxidation mixtures were dried over 3 Å molecular sieves for 16 h prior the use. This protocol (For more details see Materials and Methods, Supporting Information) was successfully applied for the preparation of oligonucleotides dT101, dT102, ON-8, and ON-9 (Table 2 and Table 3).
2.2.2. Synthetic Protocol B
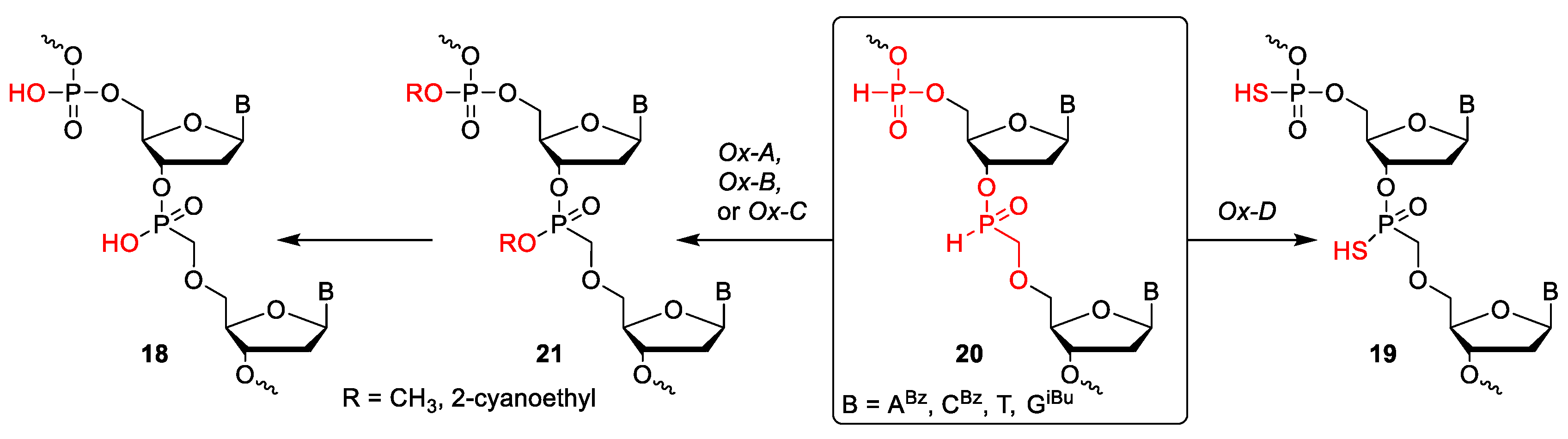
This protocol combined phosphoramidite and H-phosphonate chemistries to introduce the phosphodiester/phosphorothioate and O-methylphosphonate/O-methylphosphonothioate inter-nucleotide linkages, respectively, at specific positions of the oligonucleotide strand (Scheme 4). The PIII triester bonds were oxidized with 1M t-BuOOH in DCM or sulfurized using Sulfurizing Reagent II (DDTT, Glen Research, Sterling, VA, USA) after each phosphoramidite coupling step. The oxidation of the H-phosphinate internucleotide linkages in oligonucleotide 22 was performed after each step with the mixture Ox-C utilizing 3-hydroxypropionitrile (Table 1) to afford product 23. The use of the oxidation mixtures Ox-A and Ox-B containing methanol did not afford the desired oligonucleotide. The explanation for this behaviour might lie in the facts that neutral methyl esters of phosphonates could serve as powerful methylating agents in the presence of a nucleophile such as sulfur compound or pyridine, and that the cleavage of the methyl protecting group of the phosphonate moiety gave rise to a P–OH reactive center that interfered with the coupling steps leading to a complex mixture of products.
The synthesis of S-protected phosphonothioate internucleotide linkages in oligonucleotide 24 was achieved using the sulfur-transfer reagent (CSS) [20,21] (Table 1; Ox-E) to form S-(2-cyanoethyl) ester of the phosphonothioate linkage. After the synthesis of the entire oligonucleotide, the S-(2-cyanoethyl) ester groups were removed by β-elimination using 1 M DBU in acetonitrile. Surprisingly, gaseous ammonia could not be used for the β-elimination reaction since it caused cleavage of protected phosphonothioate internucleotide linkages to afford the degradation of the oligonucleotide strand.
The introduction of oxidation or sulfurization after each coupling step allowed the use of standard acetic anhydride-based capping procedure. The protocol (For more details see Materials and Methods, Supporting Information) was successfully applied for the preparation of oligonucleotides dT101, dT102, dT103, dT104, ON-4, ON-5, ON-6, ON-7, ON-10, and ON-11 (Table 2 and Table 3).
2.2.3. Synthetic Protocol C
This protocol combined phosphoramidite and H-phosphonate chemistries, and allowed the introduction of phosphoramidate and phosphonamidate linkages at specific sites of the oligonucleotide 25 (Scheme 5). We found that the H-phosphonate and O-methyl-(H)-phosphinate internucleotide linkages in oligonucleotide 26 were neither cleaved nor oxidized by oxaziridine CSO [24,25,26] which represented a mild phosphoramidite oxidation agent. This reagent allowed selective oxidation of PIII linkage in the presence of H-phosphonate or H-phosphinate bonds. The amidation of the P–H bond in 27 has to be performed at the end of the oligonucleotide synthesis. This synthetic arrangement did not allow the use of acetic anhydride-based capping due to the instability of H-phosphonate and H-phosphinate linkages in 27. The use of UniCap [27] could be a solution, however, in our hands we did not observe any improvement.
The amidation of P–H bonds with morpholine or N,N-dimethylenediamine in 27 (Scheme 5) was performed using CCl4/amine/DCM mixture (Table 1; Ox-F). Such mixed sequences containing P-morpholine and P–OR linkages cannot be prepared using protocol B because each detritylation step was accompanied with a partial hydrolysis of the P-morpholine bond due to the residual water content in detritylation mixture. Recently, Damha and Vlaho [28] reported the preparation of a series phosphoramidate oligonucleotides via oxidative amidation using primary amines (N,N-dimethyl-ethylendiamine, 3-(dimethylamino)propylamine, 4-dimethylaminobutylamine, isopentylamine) which were completely resistant to detritylation conditions.
This protocol (For more details see Materials and Methods, Supporting Information) was successfully applied for preparation of oligonucleotides dT103, dT104, dT105, dT106, dT107, ON-12, ON-13, ON-14, and ON-15 (Table 2 and Table 3).
2.2.4. Synthesis of Hetero-Oligonucleotides
To verify general usefulness of the above mention protocols, we also prepared a set of hetero-oligonucleotides modified at specific positions with phosphodiester/phosphonate, phosphorothioate/phosphonothioate and phosphoramidate/phosphonamidate linkages (Table 3). These oligonucleotides were prepared in excellent yields and purity. Therefore, the developed protocols represent novel and effective approaches to the synthesis of variety of phosphonate oligonucleotides modified with diverse types of linkages (For more details, and hybridization properties of ON-1–ON-15 see Supporting Information).
3. Materials and Methods
3.1 General Information
The 5′→3′ trityl-off synthesis of oligonucleotide was performed using phosphoramidite and H-phosphonate method on GeneSyn and MOS synthesizers (IOCB Prague, Prague, Czech Republic) using 3′-O-dimethoxytritylnucleoside-5′-hemisuccinate-modified LCAA-CPG and TentaGel solid supports in a 0.5 µmol scale. For the phosphoramidite chemistry all monomers, and oxidation and sulfurization agents were used according to the supplier’s recommendations (Glen Research). Condensation of H-phosphinate monomers were performed according the synthetic protocols in Table 4, Table 5, and Table 6 (for more details see Supporting Information).
3.2. H-Phosphonate Chemistry
Pyridine-acetonitrile (1:1) solutions of 0.1 M monomers (110 µL) and 0.3 M DMOCP (110 µL) were used for each coupling step (10 min). The oxidation of the oligonucleotide chain was achieved using conditions in Table 2. In case of methyl esters, the support was dried in vacuo, treated with freshly prepared thiophenol-Et3N-DMF mixture (23:32:45; v/v) for 4 h, rinsed with dry acetonitrile, dried under vacuum, and treated with gaseous NH3 (0.7 MPa) at rt overnight. In case of 2-cyanoethyl esters was thiophenol treatment omitted. The deprotected and released oligonucleotide was eluted from CPG with 0.1 M-TEAB in acetonitrile/water (1 mL; 1:1, v/v) and analyzed on a DNAPac PA100 column (Thermo Fisher Scientific, Waltham, MA, USA, 4 × 250 mm) at a flow rate of 1 mL/min using different gradients. The purification of oligonucleotide was performed on a DNAPac PA100 column (Thermo Fisher Scientific), 9 × 250 mm at a flow rate of 3 mL/min,). Pure oligonucleotide was desalted on a Luna C18 column (Phenomenex, Torrance, CA, USA, 5 μm; 10 × 50 mm;) at a flow rate of 5 mL/min using a linear gradient of acetonitrile (0→50%, 20 min) in 0.1 M aq. triethylammonium hydrogencarbonate buffer (pH 8); effluent containing oligonucleotide was evaporated to dryness on a CentriVap apparatus (Labconco, Kansas City, MO, USA) and co-evaporated with MeOH (3 × 1 mL).
4. Conclusions
In this study, we present the use of protected nucleoside-O-methyl-(H)-phosphinates as monomers for oligonucleotide synthesis using H-phosphonate chemistry. The formed O-methyl-(H)-phosphinate internucleotide linkages could be oxidized, sulfurized, or amidated similarly to the H-phosphonate bonds. We developed appropriate oxidation mixtures and three synthetic protocols allowing the combination of H-phosphonate and phosphoramidite chemistries to incorporate individual modifications either into the entire chain or at specific positions of the modified oligonucleotide. A series of DNA oligonucleotides modified with a combination of the phosphonate, phosphonothioate, phosphonamidate, phosphorothioate, phosphoramidate, and phosphodiester internucleotide linkages have been successfully synthesized to verify the robustness of the presented synthetic protocols. To our best knowledge, this is the first report of the synthesis of oligonucleotides bearing phosphonamidate and phosphonothioate internucleotide linkages. The developed protocols allow the synthesis of variety of phosphonate oligonucleotides that might be used as tools in biochemistry and biology, and in research and development of oligonucleotide-based drugs.
Supplementary Materials
The following are available online. The supplementary materials containing all experimental details.
Author Contributions
Conceptualization, O.K., O.P. and I.R.; Methodology, O.K., O.P., R.L., P.N. and I.R.; Investigation O.K., P.N., and R.L.; Analysis, O.K. and P.N.; Writing and Editing, O.K., O.P. and I.R. All authors contributed to the experiments and approved the final manuscript.
Funding
This research was funded by the Czech Science Foundation [17-12703S] to O.K., R.L., O.P. and I.R.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Šípová, H.; Špringer, T.; Rejman, D.; Šimák, O.; Petrová, M.; Novák, P.; Rosenbergová, Š.; Páv, O.; Liboska, R.; Barvík, I.; et al. 5′-O-Methylphosphonate nucleic acids—new modified DNAs that increase the Escherichia coli RNase H cleavage rate of hybrid duplexes. Nucleic Acids Res. 2014, 42, 5378–5389. [Google Scholar] [CrossRef]
- Kostov, O.; Páv, O.; Buděšínský, M.; Liboska, R.; Šimák, O.; Petrová, M.; Novák, P.; Rosenberg, I. 4-Toluenesulfonyloxymethyl-(H)-phosphinate: A reagent for the introduction of O- and S-Methyl-(H)-phosphinate Moieties. Org. Lett. 2016, 18, 2704–2707. [Google Scholar]
- Kostov, O.; Páv, O.; Rosenberg, I. Nucleoside-O-methyl-(H)-phosphinates: Novel monomers for the synthesis of methylphosphonate oligonucleotides using H-phosphonate chemistry. Curr. Protoc. Nucleic Acid Chem. 2017, 70, 4.76.1–4.76.22. [Google Scholar]
- Garegg, P.J.; Lindh, I.; Regberg, T.; Stawinski, J.; Strömberg, R.; Henrichson, C. Nucleoside H-phosphonates. III. Chemical synthesis of oligodeoxyribonucleotides by the hydrogenphosphonate approach. Tetrahedron Lett. 1986, 27, 4051–4054. [Google Scholar] [CrossRef]
- Froehler, B.C.; Ng, P.G.; Matteucci, M.D. Synthesis of DNA via deoxynudeoside H-phosphonate intermediates. Nucleic Acids Res. 1986, 14, 5399–5407. [Google Scholar] [CrossRef]
- Liu, C.; Cozens, C.; Jaziri, F.; Rozenski, J.; Maréchal, A.; Dumbre, S.; Pezo, V.; Marlière, P.; Pinheiro, V.B.; Groaz, E.; et al. Phosphonomethyl oligonucleotides as backbone-modified artificial genetic polymers. J. Am. Chem. Soc. 2018, 140, 6690–6699. [Google Scholar] [CrossRef]
- Garegg, P.J.; Regberg, T.; Stawinski, J.; Stromberg, R. Nucleoside phosphonates: Part 7. Studies on the oxidation of nucleoside phosphonate esters. J. Chem. Soc. Perkin Trans. 1 1987, 1269–1273. [Google Scholar] [CrossRef]
- Cullis, P.M.; Lee, M. The mechanism of iodine-water oxidation of H-phosphonate diesters. J. Chem. Soc. Chem. Commun. 1992, 1207–1208. [Google Scholar] [CrossRef]
- Wallin, R.; Kalek, M.; Bartoszewicz, A.; Thelin, M.; Stawinski, J. On the sulfurization of H-phosphonate diesters and phosphite triesters using elemental sulfur. PhosphorusSulfurSilicon Relat. Elem. 2009, 184, 908–916. [Google Scholar] [CrossRef]
- Mohe, N.U.; Padiya, K.J.; Salunkhe, M.M. An efficient oxidizing reagent for the synthesis of mixed backbone oligonucleotides via the H-phosphonate approach. Bioorg. Med. Chem. 2003, 11, 1419–1431. [Google Scholar] [CrossRef]
- Nilsson, J.; Stawinski, J. Oxidative coupling of H-phosphonate and H-phosphonothioate diesters. Iodine as a reagent and a catalyst. Collect. Symp. Ser. 2002, 5, 87–92. [Google Scholar]
- Nilsson, J.; Stawinski, J. Controlling stereochemistry during oxidative coupling. Preparation of Rp or Sp phosphoramidates from one P-chiral precursor. Chem. Commun. 2004, 2566–2567. [Google Scholar] [CrossRef]
- Stawinski, J.; Stromberg, R. Di- and oligonucleotide synthesis using H-phosphonate chemistry. Methods Mol. Biol. (CliftonN.J.) 2005, 288, 81–100. [Google Scholar] [CrossRef]
- Maier, M.A.; Guzaev, A.P.; Manoharan, M. Synthesis of chimeric oligonucleotides containing phosphodiester, phosphorothioate, and phosphoramidate linkages. Org. Lett. 2000, 2, 1819–1822. [Google Scholar] [CrossRef]
- Bartoszewicz, A.; Kalek, M.; Stawinski, J. The Case for the intermediacy of monomeric metaphosphate analogues during oxidation of H-phosphonothioate, H-phosphonodithioate, and H-phosphonoselenoate monoesters: Mechanistic and synthetic studies. J. Org. Chem. 2008, 73, 5029–5038. [Google Scholar] [CrossRef]
- Le Corre, S.S.; Berchel, M.; Couthon-Gourvès, H.; Haelters, J.-P.; Jaffrès, P.-A. Atherton–Todd reaction: Mechanism, scope and applications. Beilstein J. Org. Chem. 2014, 10, 1166–1196. [Google Scholar] [CrossRef]
- Letsinger, R.L.; Singman, C.N.; Histand, G.; Salunkhe, M. Cationic oligonucleotides. J. Am. Chem. Soc. 1988, 110, 4470–4471. [Google Scholar] [CrossRef]
- Jung, P.M.; Histand, G.; Letsinger, R.L. Hybridization of alternating cationic/anionic oligonucleotides to RNA segments. Nucleosides Nucleotides 1994, 13, 1597–1605. [Google Scholar] [CrossRef]
- Paul, S.; Roy, S.; Monfregola, L.; Shang, S.; Shoemaker, R.; Caruthers, M.H. Oxidative substitution of boranephosphonate diesters as a route to post-synthetically modified DNA. J. Am. Chem. Soc. 2015, 137, 3253–3264. [Google Scholar] [CrossRef]
- Brill, W.K.D. Thioalkylation of nucleoside-H-phosphonates and its application to solid phase synthesis of oligonucleotides. Tetrahedron Lett. 1995, 36, 703–706. [Google Scholar] [CrossRef]
- Reese, C.B.; Yan, H. Solution phase synthesis of ISIS 2922 (Vitravene) by the modified H-phosphonate approach. J. Chem. Soc. Perkin Trans. 1 2002, 2619–2633. [Google Scholar] [CrossRef]
- Alefelder, S.; Patel, B.K.; Eckstein, F. Incorporation of terminal phosphorothioates into oligonucleotides. Nucleic Acids Res. 1998, 26, 4983–4988. [Google Scholar] [CrossRef] [Green Version]
- Andrus, A.; Efcavitch, J.W.; McBride, L.J.; Giusti, B. Novel activating and capping reagents for improved hydrogen-phosphonate DNA synthesis. Tetrahedron Lett. 1988, 29, 861–864. [Google Scholar] [CrossRef]
- Ugi, I.; Jacob, P.; Landgraf, B.; Ruppf, C.; Lemmen, P.; Verfürth, U. Phosphite oxidation and the preparation of five-membered cyclic phosphorylating reagents via the phosphites. Nucleosides Nucleotides 1988, 7, 605–608. [Google Scholar] [CrossRef]
- Reynolds, M.A.; Hogrefe, R.I.; Jaeger, J.A.; Schwartz, D.A.; Riley, T.A.; Marvin, W.B.; Daily, W.J.; Vaghefi, M.M.; Beck, T.A.; Knowles, S.K.; et al. Synthesis and thermodynamics of oligonucleotides containing chirally pure R P methylphosphonate linkages. Nucleic Acids Res. 1996, 24, 4584–4591. [Google Scholar] [CrossRef]
- Dellinger, D.J.; Sheehan, D.M.; Christensen, N.K.; Lindberg, J.G.; Caruthers, M.H. Solid-phase chemical synthesis of phosphonoacetate and thiophosphonoacetate oligodeoxynucleotides. J. Am. Chem. Soc. 2003, 125, 940–950. [Google Scholar] [CrossRef]
- Glen Research Unicap Phosphoramidite, An Alternative to Acetic Anhydride Capping. Available online: http://www.glenresearch.com/GlenReports/GR17-13.html (accessed on 9 October 2018).
- Vlaho, D.; Damha, M.J. Synthesis of chimeric oligonucleotides having modified internucleotide linkages via an automated H-phosphonate/phosphoramidite approach. Curr. Protoc. Nucleic Acid Chem. 2018, 73, e53. [Google Scholar] [CrossRef]
Sample Availability: Not available. |
Figure 1.
Overview of possible combinations of the phosphodiester 3, 4, 6 and phosphonate 8a, 8b, 9a, 9b and 10 linkages.
Figure 1.
Overview of possible combinations of the phosphodiester 3, 4, 6 and phosphonate 8a, 8b, 9a, 9b and 10 linkages.

Scheme 1.
Model dimer for the optimization of H-phosphonate chemistry.

Scheme 2.
Stability of phosphorothioate/phosphonothioate and phosphoramidate/phosphonamidate internucleotide linkages.
Scheme 2.
Stability of phosphorothioate/phosphonothioate and phosphoramidate/phosphonamidate internucleotide linkages.

Scheme 3.
Synthetic protocol A—synthesis of fully modified strands.

Scheme 4.
Synthetic protocol B—synthesis of oligonucleotides modified at specific positions.

Scheme 5.
Synthetic protocol C—synthesis of phosphoramidate/phosphonamidate strands.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The conditions of oxidation, sulfurization and amidation.
| Code | Mixture | Oxidation Time (min) | |
|---|---|---|---|
| H-Phosphonate | H-Phosphinate | ||
| Ox-A | CCl4/MeOH/MeIm (7:2:1) | 15 | 30 |
| Ox-B | CCl4/MeOH/MeIm/Et3N (7:2:0.5:0.5) | 10 | 20 |
| Ox-C | 0.1 M HOCH2CH2CN in CCl4/DCM/MeIm (6:2:2) | 15 | 30 |
| Ox-D | 0.5 M sulfur in pyridine | 20 | 20 |
| Ox-E | 0.2 M CSS in ACN/BSTFA (29:1) | 30 | 30 |
| Ox-F | CCl4/amine */DCM (3:2:5) | 180 | 180 |
* Morpholine or N,N-dimethylethylendiamine.
Table 2.
Summary of model homo-oligothymidylates dT101–dT107.
| Internucleotide Linkages | Code | Sequence 5′→3′ | Synthetic Protocol (HPLC Purity) | MALDI TOF Calcd Found | |
|---|---|---|---|---|---|
 | dT101 | d(T T T T T T T T T T) | A (70%), B (74%), C (64%) | 3020.5516 | 3020.5733 |
| dT102 | d(T T T T T T T T T T) | A (59%), B (77%) | 3164.3460 | 3164.3219 | |
 | dT103 | d(T T T T T T T T T T) | B (36%), C (88%) | 3187.3049 | 3187.1703 |
| dT104 | d(T T T T T T T T T T) | B (42%), C (70%) | 3229.3859 | 3229.1063 | |
 | dT105 | d(T T T T T T T T T T) | C (30%) | 3070.2479 | 3070.7329 |
| dT106 | d(T T T T T T T T T T) | C (75%) | 2979.9839 | 2980.3970 | |
| dT107 | d(T T T T T T T T T T) | C (77%) | 3028.1696 | 3028.4048 | |
Table 3.
Summary of hetero-oligonucleotides ON-1–ON-15.
| Internucleotide Linkages | Code | Sequence 5′→3′ | Synthetic Protocol (HPLC Purity) | MALDI TOF | |
|---|---|---|---|---|---|
| Calcd | Found | ||||
 | ON-1 | d(G C A T A T C A C) | B (86%) | 2729.4322 | 2729.5331 |
| ON-2 | d(G C A T A T C A C) | B (84%) | 2761.3865 | 2761.1589 | |
| ON-3 | d(G C A T A T C A C) | B (81%) | 2809.3180 | 2809.3292 | |
 | ON-4 | d(G C A T A T C A C) | B (21%) | 2771.4792 | 2771.3983 |
| ON-5 | d(G C A T A T C A C) | B (56%) | 2899.2964 | 2899.2376 | |
| ON-6 | d(G C A T A T C A C) | B (51%) | 2819.4106 | 2819.2887 | |
| ON-7 | d(G C A T A T C A C) | B (43%) | 2851.3650 | 2851.3559 | |
 | ON-8 | d(G C A T A T C A C) | A (76%) | 2723.5477 | 2723.7076 |
| ON-9 | d(G C A T A T C A C) | A (84%) | 2851.3650 | 2851.2513 | |
| ON-10 | d(G C A T A T C A C) | B (86%) | 2771.4791 | 2771.3636 | |
| ON-11 | d(G C A T A T C A C) | B (86%) | 2803.4335 | 2803.5659 | |
 | ON-12 | d(G C A T A T C A C) | C (59%) | 2930.7213 | 2930.6797 |
| ON-13 | d(G C A T A T C A C) | C (66%) | 2975.8631 | 2975.7741 | |
| ON-14 | d(G C A T A T C A C) | C (78%) | 2888.6743 | 2888.8365 | |
| ON-15 | d(G C A T A T C A C) | C (79%) | 2933.8162 | 2933.7591 | |
Table 4.
Synthetic Protocol A (For more details see Supporting Information).
Table 4.
Synthetic Protocol A (For more details see Supporting Information).
| Procedure | Reagents | Volume/Time |
|---|---|---|
| H-Phosphonate/H-Phosphinate Condensation Step | ||
| Deblocking | 3% DCA in DCM | Flow through the column 6 mL/2 min |
| Condensation | 0.1M monomer in ACN/Py (1:1) | 120 µL/10min |
| 0.3M DMOCP in ACN/Py (95:5) | ||
| Oxidation | Ox-A, Ox-B, Ox-D | 220 µL/30 min |
| Methyl-ester cleavage * | PhSH/Et3N/DMF(1:1.4:2) | 300 µL/4 h |
| Cleavage from solid support | Gaseous ammonia at 0.7 MPa | Gas chamber/16 h |
* Applicable only in case of Ox-A and Ox-B mixtures containing methanol.
Table 5.
Synthetic Protocol B (For more details see Supporting Information).
Table 5.
Synthetic Protocol B (For more details see Supporting Information).
| Procedure | Reagents | Volume/Time |
|---|---|---|
| H-Phosphonate/H-Phosphinate Condensation Step | ||
| Deblocking | 3% DCA in DCM | Flow through the column 6 mL/2 min |
| DCA washout | 5% pyridine in methanol | |
| Condensation | 0.1M monomer in ACN/Py (1:1) | 120 µL/10 min |
| 0.3M DMOCP in ACN/Py (95:5) | ||
| Oxidation or Sulfurization | Ox-C or Ox-E | 220 µL/30 min |
| Capping | Ac2O/Py/THF (1:1:1:17) | 2 × 150 µL/3 min |
| MeIm/THF (1:9) | ||
| Phosphoramidite Condensation Step | ||
| Deblocking | 3% DCA in DCM | Flow through the column 6 mL/2 min |
| DCA washout | 5% pyridine in methanol | |
| Condensation | 0.1M monomer in ACN | 120 µL/5 min |
| 0.25M ETT in ACN | ||
| Oxidation or Sulfurization | CSO, tBuOOH or DDTT | 220 µL/3.5 min |
| End of Synthetic Cycle | ||
| S-cyanoethyl-ester cleavage * | 1M DBU in ACN | 220 µL/3 min |
| Cleavage from solid support | Ammonia gas at 0.7 MPa | Gas chamber/16 h |
* Applicable only in case of CSS sulfurization mixtures.
Table 6.
Synthetic Protocol C (For more details see Supporting Information).
Table 6.
Synthetic Protocol C (For more details see Supporting Information).
| Procedure | Reagents | Volume/Time |
|---|---|---|
| H-Phosphonate/H-Phosphinate Condensation Steps | ||
| Deblocking | 3% DCA in DCM | Flow through the column 6 mL/2 min |
| Condensation | 0.1 M monomer in ACN/Py (1:1) | 120 µL/10 min |
| 0.3 M DMOCP in ACN/Py (95:5) | ||
| Phosphoramidite Condensation Step | ||
| Deblocking | 3% DCA in DCM | Flow through the column 6 mL/2 min |
| DCA washout | 5% pyridine in methanol | |
| Condensation | 0.1 M monomer in ACN | 120 µL/5 min |
| 0.25 M ETT in ACN | ||
| Oxidation | CSO | 220 µL/3.5 min |
| End of Synthetic Cycle | ||
| Amidation of P–H bond | Ox-F | 220 µL/180 min |
| Cleavage from solid support | Ammonia gas at 0.7 MPa | Gas chamber/16 h |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kostov, O.; Liboska, R.; Páv, O.; Novák, P.; Rosenberg, I. Solid-Phase Synthesis of Phosphorothioate/Phosphonothioate and Phosphoramidate/Phosphonamidate Oligonucleotides. Molecules 2019, 24, 1872. https://doi.org/10.3390/molecules24101872
AMA Style
Kostov O, Liboska R, Páv O, Novák P, Rosenberg I. Solid-Phase Synthesis of Phosphorothioate/Phosphonothioate and Phosphoramidate/Phosphonamidate Oligonucleotides. Molecules. 2019; 24(10):1872. https://doi.org/10.3390/molecules24101872
Chicago/Turabian StyleKostov, Ondřej, Radek Liboska, Ondřej Páv, Pavel Novák, and Ivan Rosenberg. 2019. "Solid-Phase Synthesis of Phosphorothioate/Phosphonothioate and Phosphoramidate/Phosphonamidate Oligonucleotides" Molecules 24, no. 10: 1872. https://doi.org/10.3390/molecules24101872