A Flexible Synthesis of 68Ga-Labeled Carbonic Anhydrase IX (CAIX)-Targeted Molecules via CBT/1,2-Aminothiol Click Reaction

Abstract
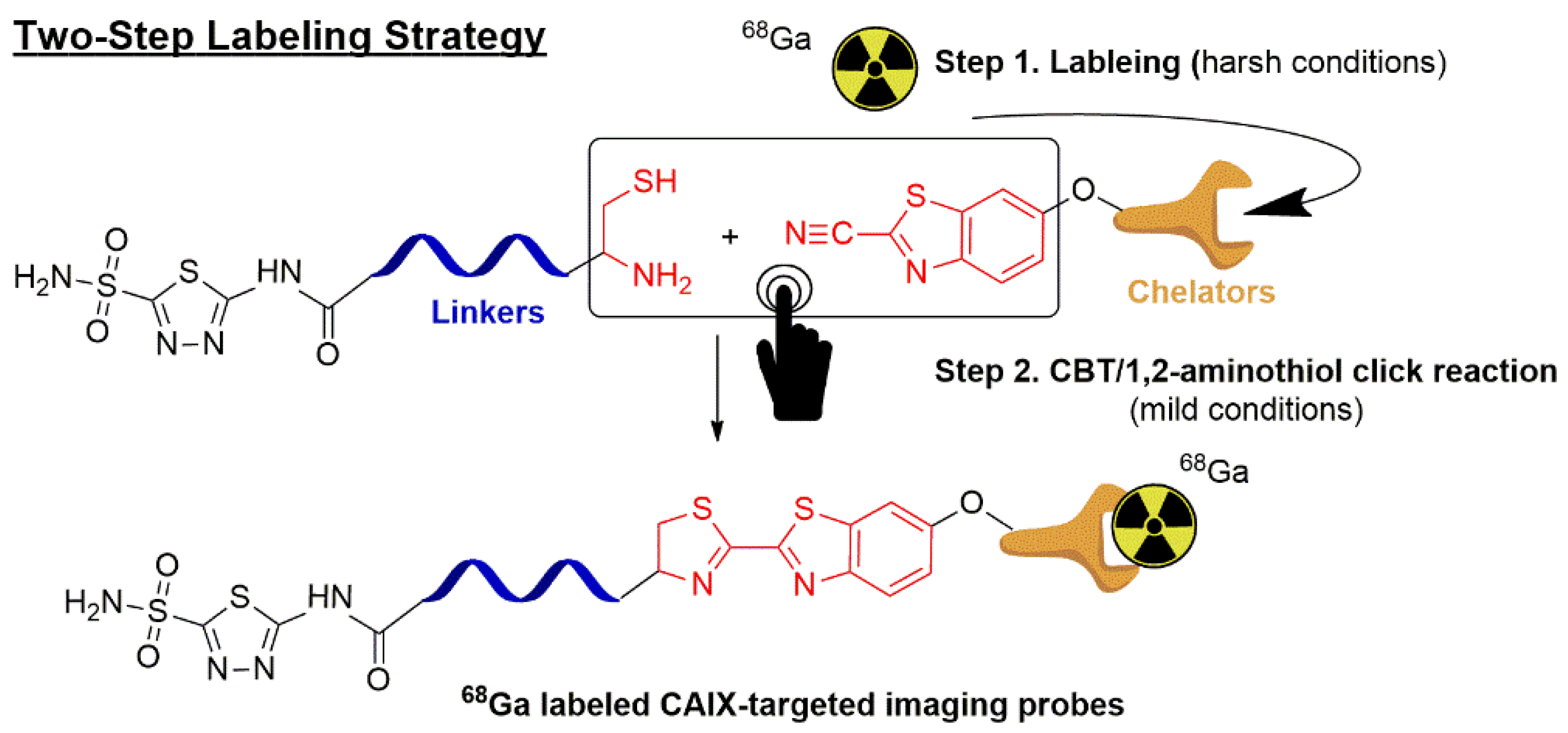
:1. Introduction
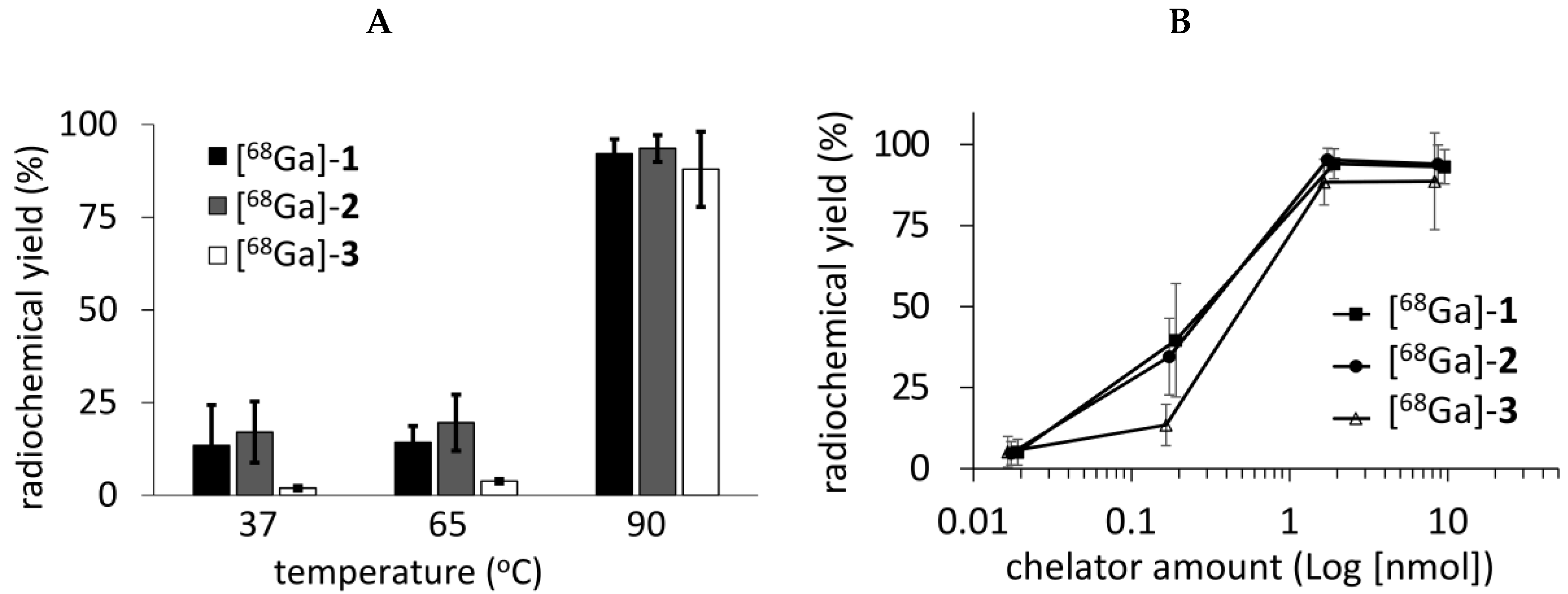
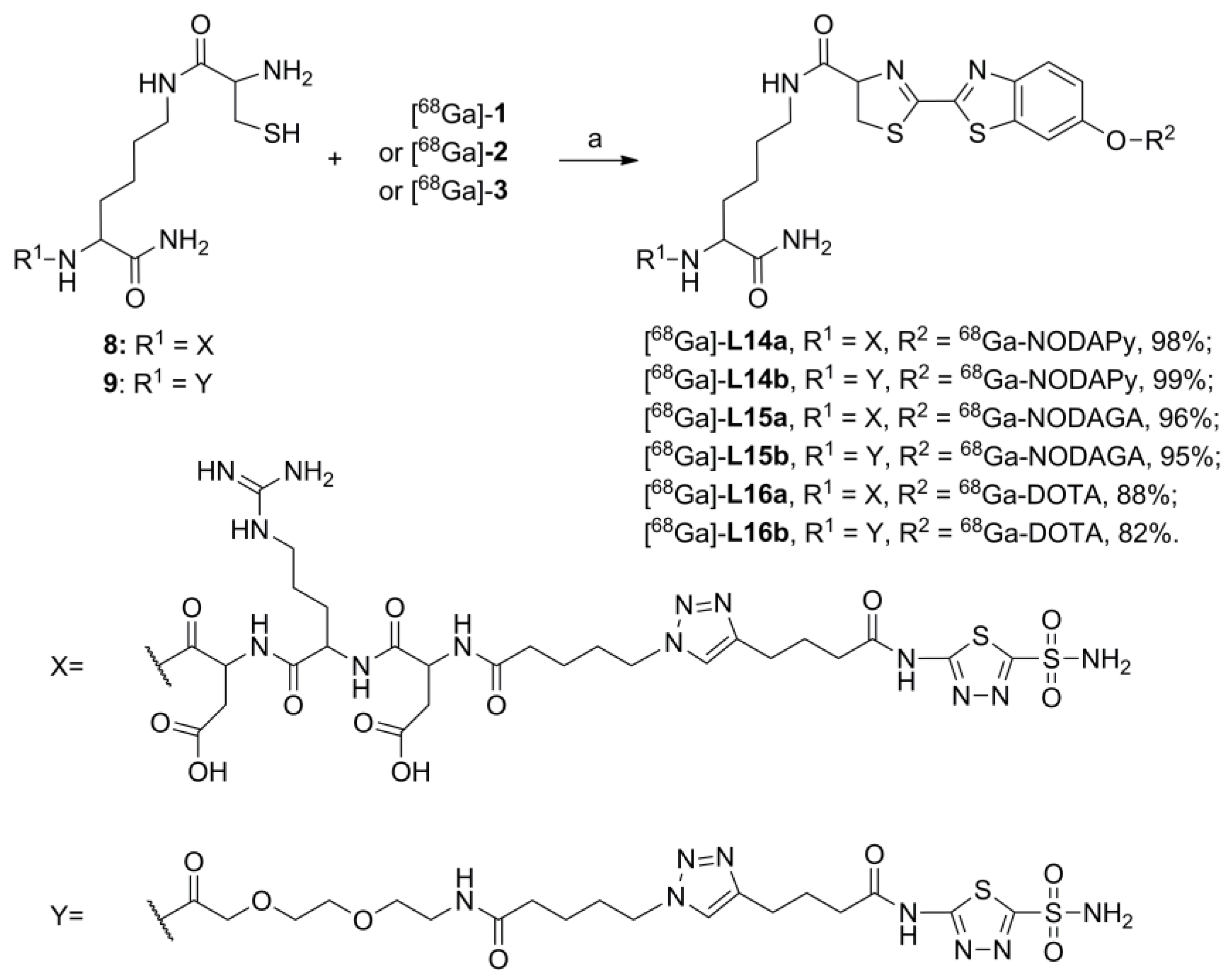
2. Results and Discussions
3. Materials and Methods
3.1. General Information
3.2. High-Performance Liquid Chromatography (HPLC) Conditions for Analysis
3.3. HPLC Conditions for Purification
3.4. Chemistry and Radiolabeling
3.5. Determination of LogD7.4
3.6. Stability and Challenge Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tanzey, S.S.; Thompson, S.; Scott, P.J.; Brooks, A.F. Gallium-68: Methodology and novel radiotracers for positron emission tomography (2012–2017). Pharm. Pat. Anal. 2018, 7, 193–227. [Google Scholar] [CrossRef] [PubMed]
- Notni, J.; Simecek, J.; Hermann, P.; Wester, H.J. TRAP, a powerful and versatile framework for gallium-68 radiopharmaceuticals. Chem. Eur. J. 2011, 17, 14718–14722. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.L.; Carroll, L.; Aboagye, E.O.; Spivey, A.C. Bioorthogonal chemistry for (68) Ga radiolabelling of DOTA-containing compounds. J. Labelled Comp. Radiopharm. 2014, 57, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, A.; Läppchen, T.; Weinmann, C.; Bier-Schorr, L.; Keller, M.; Kiefer, Y.; Holland, J.P.; Bartholomä, M.D. Gallium Complexation, Stability, and Bioconjugation of 1,4,7-Triazacyclononane Derived Chelators with Azaheterocyclic Arms. Inorg. Chem. 2017, 56, 9097–9110. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Erler, J. Hypoxia-mediated metastasis. Adv. Exp. Med. Biol. 2014, 772, 55–81. [Google Scholar] [CrossRef]
- McDonald, P.C.; Winum, J.Y.; Supuran, C.T.; Dedhar, S. Recent developments in targeting carbonic anhydrase IX for cancer therapeutics. Oncotarget 2012, 3, 84–97. [Google Scholar] [CrossRef]
- Sedlakova, O.; Svastova, E.; Takacova, M.; Kopacek, J.; Pastorek, J.; Pastorekova, S. Carbonic anhydrase IX, a hypoxia-induced catalytic component of the pH regulating machinery in tumors. Front. Physiol. 2014, 4, 400. [Google Scholar] [CrossRef]
- Swietach, P.; Hulikova, A.; Vaughan-Jones, R.D.; Harris, A.L. New insights into the physiological role of carbonic anhydrase IX in tumour pH regulation. Oncogene 2010, 29, 6509–6521. [Google Scholar] [CrossRef] [Green Version]
- Lou, Y.; McDonald, P.C.; Oloumi, A.; Chia, S.; Ostlund, C.; Ahmadi, A.; Kyle, A.; Auf dem Keller, U.; Leung, S.; Huntsman, D.; et al. Targeting tumor hypoxia: Suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res. 2011, 71, 3364–3376. [Google Scholar] [CrossRef]
- Ilie, M.; Mazure, N.M.; Hofman, V.; Ammadi, R.E.; Ortholan, C.; Bonnetaud, C.; Havet, K.; Venissac, N.; Mograbi, B.; Mouroux, J.; et al. High levels of carbonic anhydrase IX in tumour tissue and plasma are biomarkers of poor prognostic in patients with non-small cell lung cancer. Br. J. Cancer 2010, 102, 1627–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klatte, T.; Seligson, D.B.; Rao, J.Y.; Yu, H.; de Martino, M.; Kawaoka, K.; Wong, S.G.; Belldegrun, A.S.; Pantuck, A.J. Carbonic anhydrase IX in bladder cancer: A diagnostic, prognostic, and therapeutic molecular marker. Cancer 2009, 115, 1448–1458. [Google Scholar] [CrossRef]
- Choschzick, M.; Oosterwijk, E.; Müller, V.; Woelber, L.; Simon, R.; Moch, H.; Tennstedt, P. Overexpression of carbonic anhydrase IX (CAIX) is an independent unfavorable prognostic marker in endometrioid ovarian cancer. Virchows. Arch. 2011, 459, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.Y.; Aurelio, O.N.; Jan, K.; Zavada, J.; Stanbridge, E.J. Identification of the MN/CA9 protein as a reliable diagnostic biomarker of clear cell carcinoma of the kidney. Cancer Res. 1997, 57, 2827–2831. [Google Scholar] [CrossRef] [PubMed]
- Krall, N.; Pretto, F.; Mattarella, M.; Muller, C.; Neri, D. A 99mTc-Labeled ligand of carbonic anhydrase IX selectively targets renal cell carcinoma in vivo. J. Nucl. Med. 2016, 57, 943–949. [Google Scholar] [CrossRef]
- Zhang, Z.; Lau, J.; Zhang, C.; Colpo, N.; Nocentini, A.; Supuran, C.T.; Bénard, F.; Lin, K.S. Design, synthesis and evaluation of (18)F-labeled cationic carbonic anhydrase IX inhibitors for PET imaging. J. Enzyme Inhib. Med. Chem. 2017, 32, 722–730. [Google Scholar] [CrossRef]
- Pan, J.; Lau, J.; Mesak, F.; Hundal, N.; Pourghiasian, M.; Liu, Z.; Bénard, F.; Dedhar, S.; Supuran, C.T.; Lin, K.S. Synthesis and evaluation of 18F-labeled carbonic anhydrase IX inhibitors for imaging with positron emission tomography. J. Enzyme Inhib. Med. Chem. 2014, 29, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.; Zhang, Z.; Jenni, S.; Kuo, H.T.; Liu, Z.; Vullo, D.; Supuran, C.T.; Lin, K.S.; Bénard, F. PET Imaging of Carbonic Anhydrase IX Expression of HT-29 Tumor Xenograft Mice with (68)Ga-Labeled Benzenesulfonamides. Mol. Pharm. 2016, 13, 1137–1146. [Google Scholar] [CrossRef]
- Sneddon, D.; Niemans, R.; Bauwens, M.; Yaromina, A.; van Kuijk, S.J.; Lieuwes, N.G.; Biemans, R.; Pooters, I.; Pellegrini, P.A.; Lengkeek, N.A.; et al. Synthesis and in vivo biological Evaluation of (68)Ga-Labeled Carbonic Anhydrase IX Targeting Small Molecules for Positron Emission Tomography. J. Med. Chem. 2016, 59, 6431–6443. [Google Scholar] [CrossRef]
- Mueller, D.; Breeman, W.A.; Klette, I.; Gottschaldt, M.; Odparlik, A.; Baehre, M.; Tworowska, I.; Schultz, M.K. Radiolabeling of DOTA-like conjugated peptides with generator-produced (68)Ga and using NaCl-based cationic elution method. Nat. Protoc. 2016, 11, 1057–1066. [Google Scholar] [CrossRef]
- Velikyan, I. Prospective of (68)Ga-radiopharmaceutical development. Theranostics 2013, 4, 47–80. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhang, C.J.; Chen, G.; Na, Z.; Yao, S.Q.; Sun, H. Site-specific immobilization of biomolecules by a biocompatible reaction between terminal cysteine and 2-cyanobenzothiazole. Chem. Commun. 2013, 49, 8644–8646. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Xiao, F.; Zhan, K.; Kim, Y.P.; Xie, H.; Xia, Z.; Rao, J. A biocompatible condensation reaction for the labeling of terminal cysteine residues on proteins. Angew. Chem. Int. Ed. Engl. 2009, 48, 9658–9662. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liang, G. Applications of CBT-Cys click reaction: Past, present, and future. Sci. China Chem. 2018, 61, 1088–1098. [Google Scholar] [CrossRef]
- Chen, K.T.; Ieritano, C.; Seimbille, Y. Early-Stage Incorporation Strategy for Regioselective Labeling of Peptides using the 2-Cyanobenzothiazole/1,2-Aminothiol Bioorthogonal Click Reaction. Chem. Open 2018, 7, 256–261. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.; Shen, B.; Xiong, L.; Miao, Z.; Lee, K.H.; Rao, J.; Chin, F.T. Efficient method for site-specific 18F-labeling of biomolecules using the rapid condensation reaction between 2-cyanobenzothiazole and cysteine. Bioconjug. Chem. 2012, 23, 1902–1908. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Ieritano, C.; Chen, K.T.; Dias, G.M.; Rousseau, J.; Bénard, F.; Seimbille, Y. Two bifunctional desferrioxamine chelators for bioorthogonal labeling of biovectors with zirconium-89. Org. Biomol. Chem. 2018, 16, 5102–5106. [Google Scholar] [CrossRef] [PubMed]
- Kubícek, V.; Havlícková, J.; Kotek, J.; Tircsó, G.; Hermann, P.; Tóth, E.; Lukes, I. Gallium(III) complexes of DOTA and DOTA-monoamide: Kinetic and thermodynamic studies. Inorg. Chem. 2010, 49, 10960–10969. [Google Scholar] [CrossRef]
- Clarke, E.T.; Martell, A.E. Stabilities of the Fe(III), Ga(III) and In(III) chelates of N,N′,N″-triazacyclononanetriacetic acid. Inorganica Chim. Acta 1991, 181, 273–280. [Google Scholar] [CrossRef]
- Chen, K.; Chen, X. Design and development of molecular imaging probes. Curr. Top. Med. Chem. 2010, 10, 1227–1236. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | 8 (nmol) | Buffer pH | Time (min) | RCY (%) a |
|---|---|---|---|---|
| 1 | 10 | 7.4 | 30 | 54 |
| 2 | 10 | 7.4 | 60 | 78 |
| 3 | 10 | 7.4 | 180 | 98 |
| 4 | 10 | 9.0 | 30 | 85 |
| 5 | 10 | 9.0 | 60 | 99 |
| 6 | 25 | 9.0 | 20 | 99 |
| Compound | Stability (%) a | Log D7.4 |
|---|---|---|
| [68Ga]-L14a | >99 | −1.95 ± 0.03 |
| [68Ga]-L14b | >99 | −2.03 ± 0.10 |
| [68Ga]-L15a | >99 | −2.68 ± 0.02 |
| [68Ga]-L15b | >99 | −2.71 ± 0.05 |
| [68Ga]-L16a | >99 | −3.20 ± 0.06 |
| [68Ga]-L16b | >99 | −3.29 ± 0.14 |
| 68Ga-Labeled BFCs | AAZs | Products | RCP (%) | tR (min)/ HPLC System |
|---|---|---|---|---|
| [68Ga]-1 | 8 | [68Ga]-L14a | 98 | 9.4 / A |
| [68Ga]-1 | 9 | [68Ga]-L14b | 99 | 10.3 / A |
| [68Ga]-2 | 8 | [68Ga]-L15a | 96 | 9.9 / A |
| [68Ga]-2 | 9 | [68Ga]-L15b | 95 | 10.8 / A |
| [68Ga]-3 | 8 | [68Ga]-L16a | 88 | 9.5 / A |
| [68Ga]-3 | 9 | [68Ga]-L16b | 82 | 18.5 / B * |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, K.-T.; Nguyen, K.; Ieritano, C.; Gao, F.; Seimbille, Y. A Flexible Synthesis of 68Ga-Labeled Carbonic Anhydrase IX (CAIX)-Targeted Molecules via CBT/1,2-Aminothiol Click Reaction. Molecules 2019, 24, 23. https://doi.org/10.3390/molecules24010023
Chen K-T, Nguyen K, Ieritano C, Gao F, Seimbille Y. A Flexible Synthesis of 68Ga-Labeled Carbonic Anhydrase IX (CAIX)-Targeted Molecules via CBT/1,2-Aminothiol Click Reaction. Molecules. 2019; 24(1):23. https://doi.org/10.3390/molecules24010023
Chicago/Turabian StyleChen, Kuo-Ting, Kevin Nguyen, Christian Ieritano, Feng Gao, and Yann Seimbille. 2019. "A Flexible Synthesis of 68Ga-Labeled Carbonic Anhydrase IX (CAIX)-Targeted Molecules via CBT/1,2-Aminothiol Click Reaction" Molecules 24, no. 1: 23. https://doi.org/10.3390/molecules24010023