
Self-Assembly of Short Elastin-like Amphiphilic Peptides: Effects of Temperature, Molecular Hydrophobicity and Charge Distribution


Abstract
:1. Introduction
2. Experimental
2.1. Materials and Solution Preparation
2.2. Fluorescence Measurement
2.3. Atomic Force Microscopy (AFM)
2.4. Transmission Electron Microscopy (TEM)
2.5. Circular Dichroism (CD)
2.6. Fourier Transform Infrared Spectroscopy (FTIR)
3. Results and Discussion
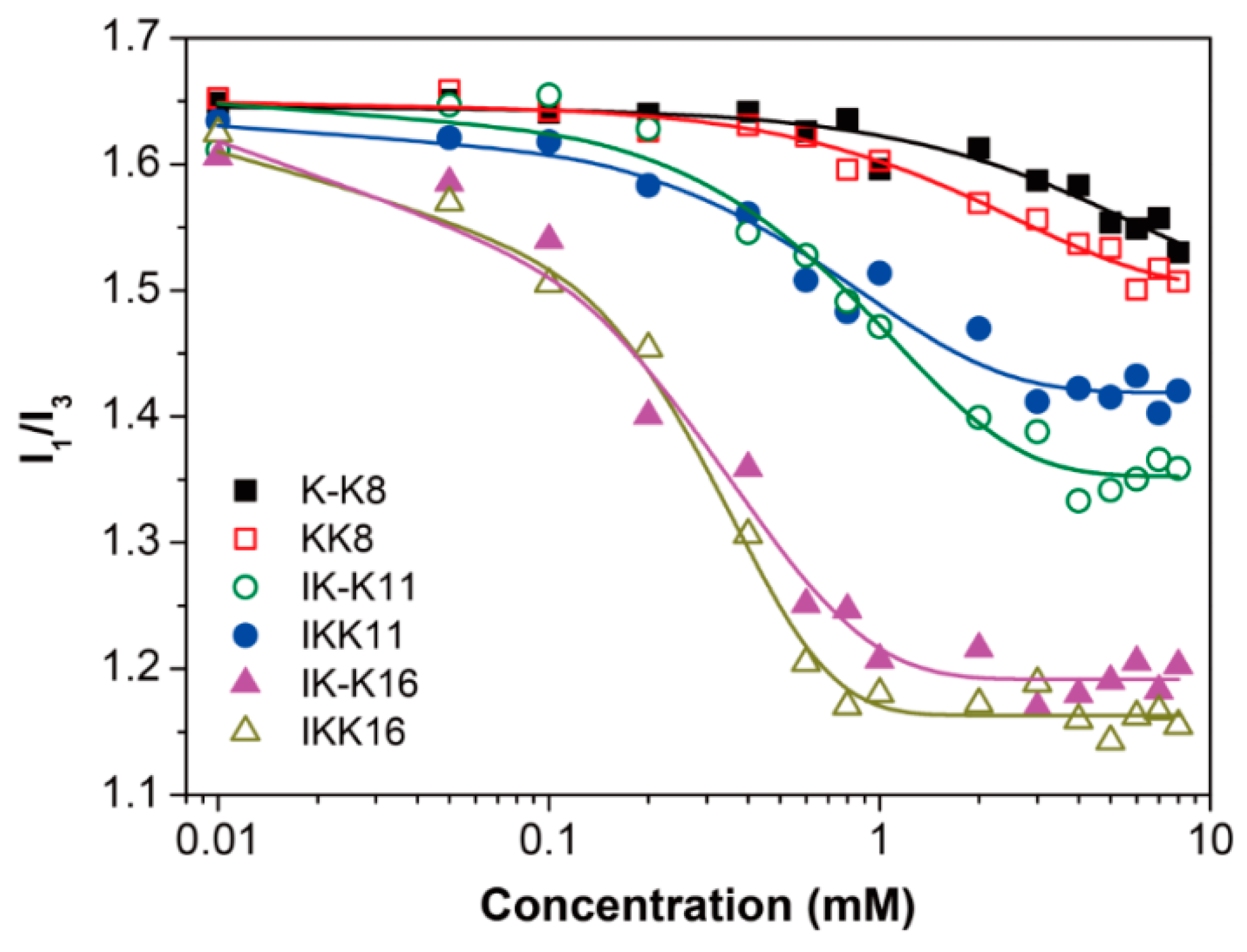
3.1. Determination of CACs
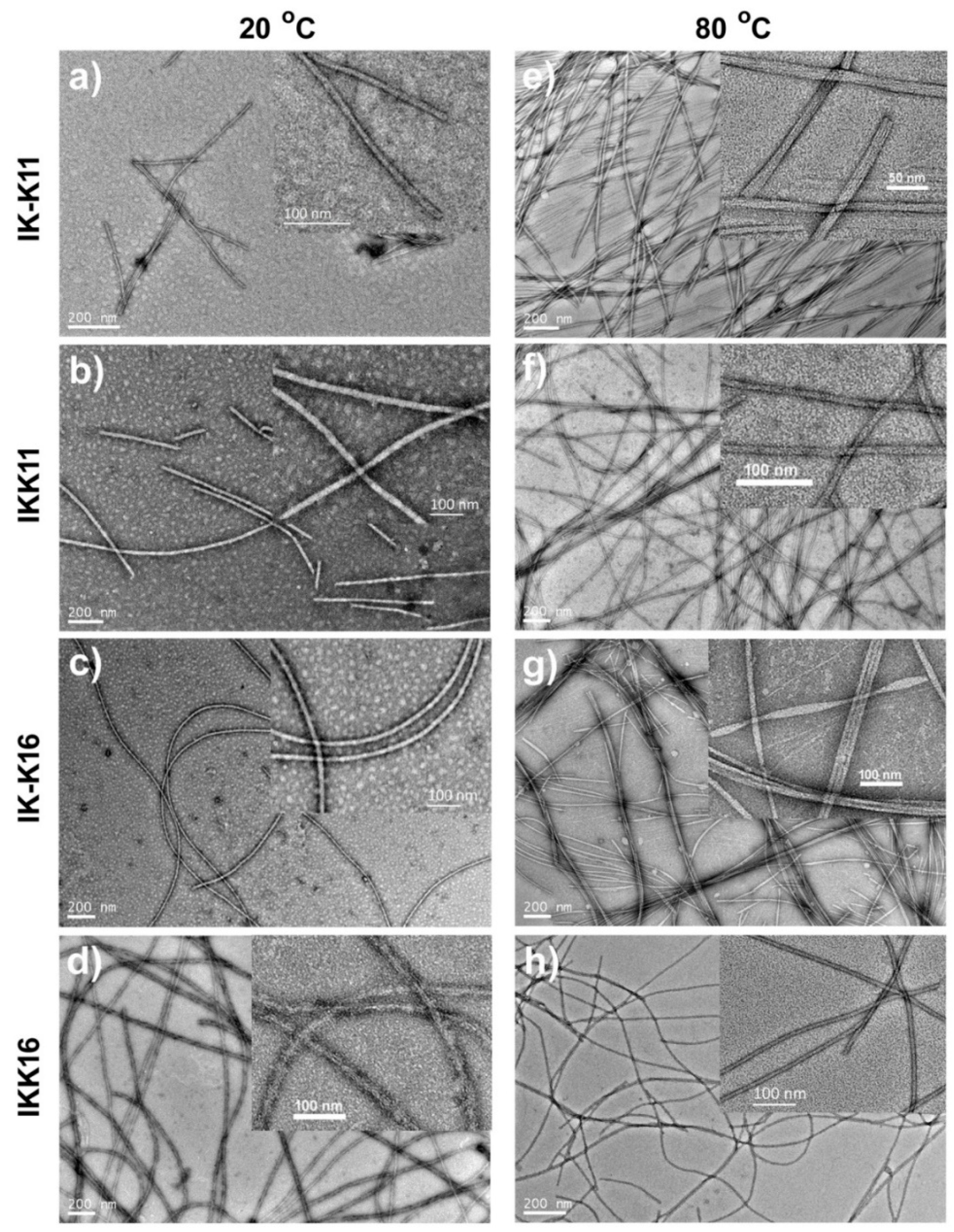
3.2. Morphologies of the Self-Assembled Structures
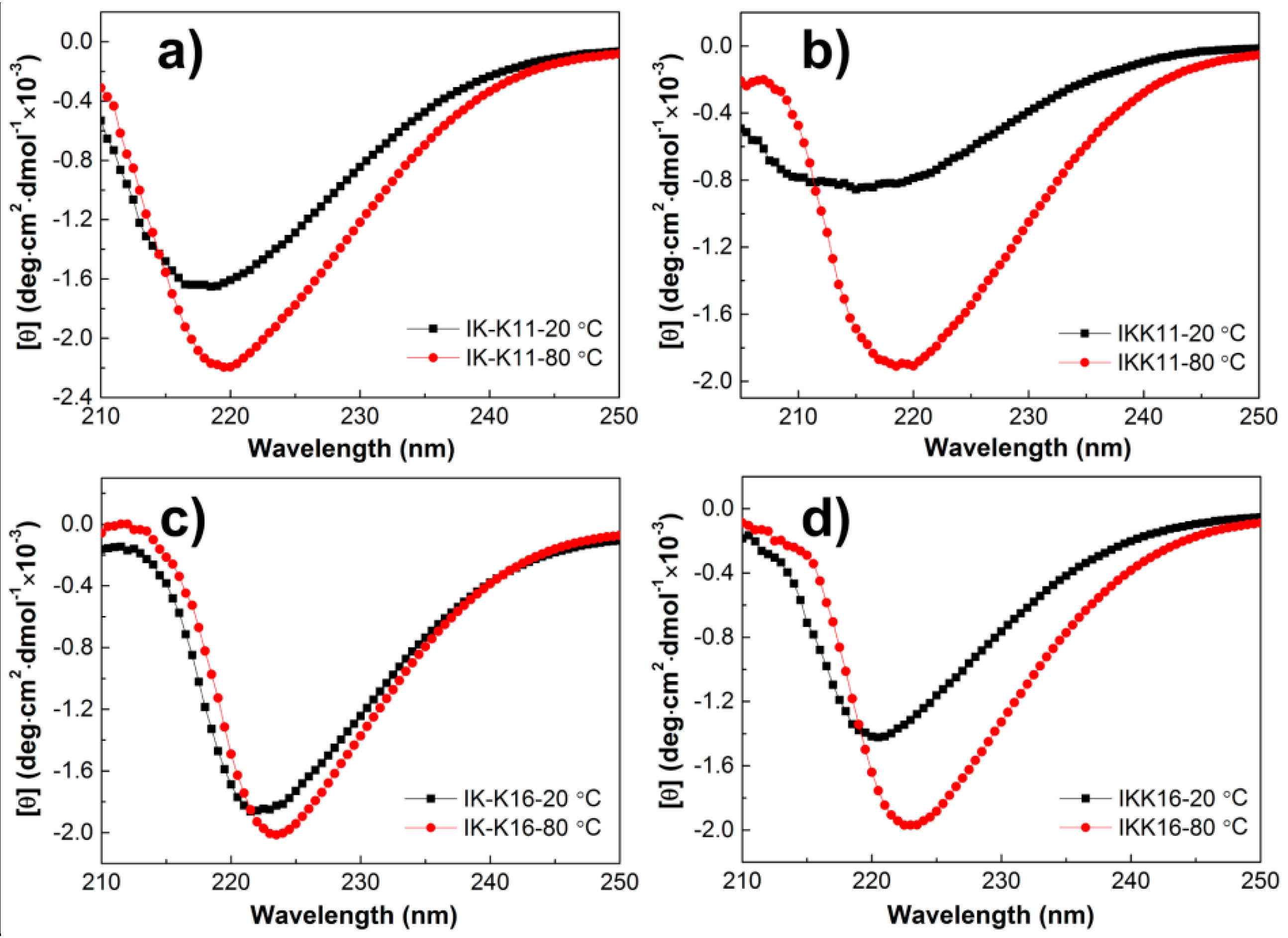
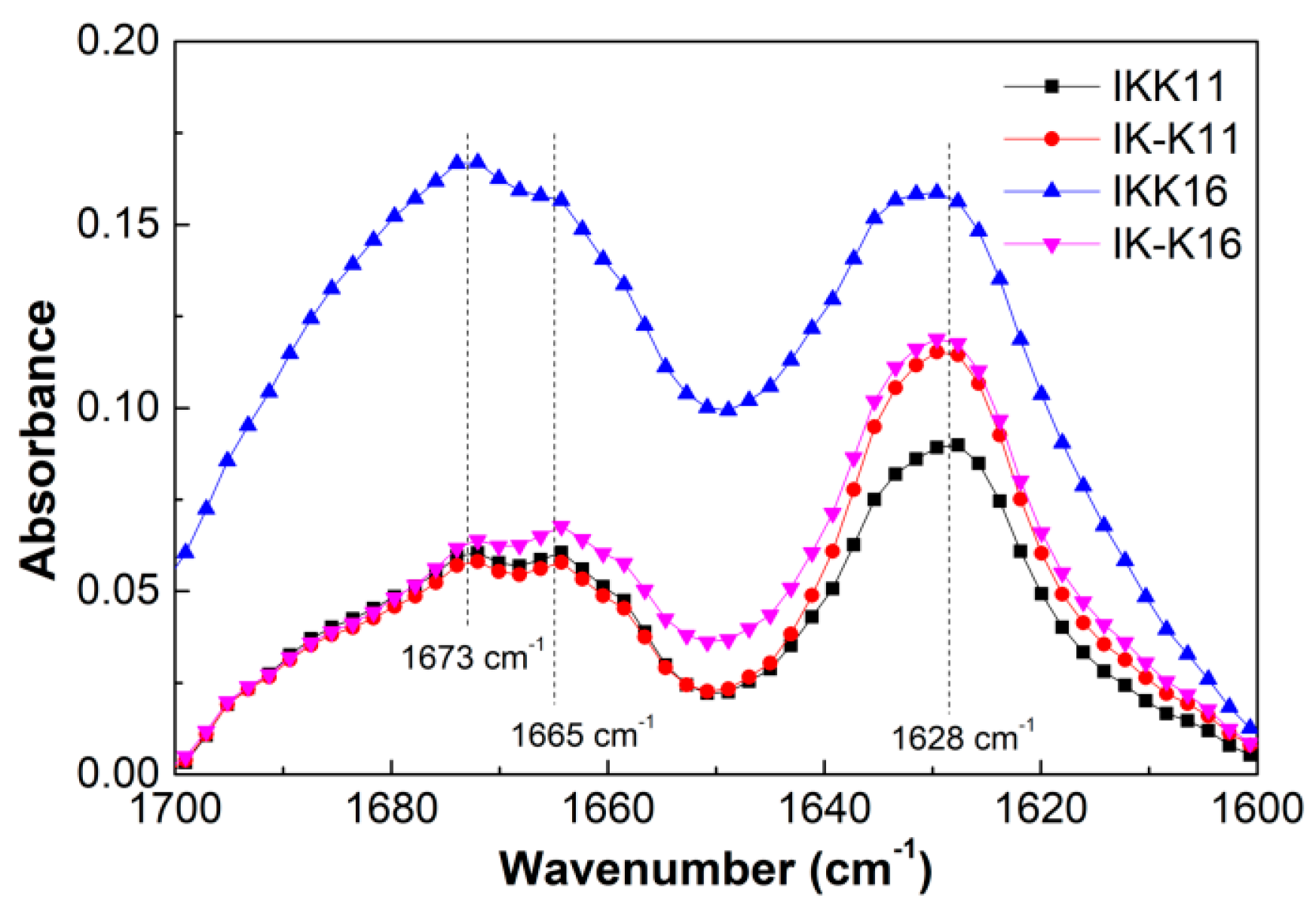
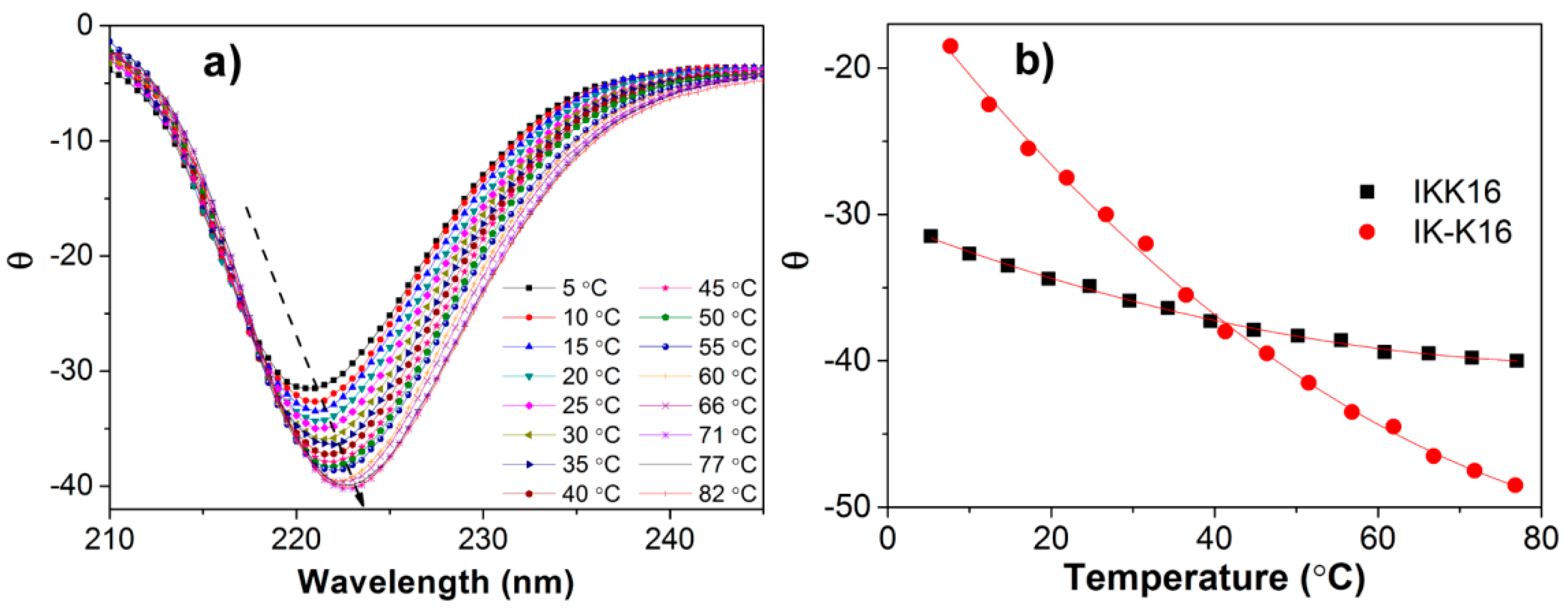
3.3. Secondary Structures at Varied Temperatures
3.4. Effects of Molecular Hydrophobicity, Charge Distribution and Temperature on Peptide Self-assembly
4. Conclusions
Supporting Information
Author Contributions
Funding
Conflicts of Interest
References
- Urry, D.W. Physical chemistry of biological free energy transduction as demonstrated by elastic protein-based polymers. J. Phys. Chem. B 1997, 101, 11007–11028. [Google Scholar] [CrossRef]
- Mithieux, S.M.; Weiss, A.S. Elastin. In Advances in Protein Chemistry; Elsevier Inc.: Amsterdam, The Netherlands, 2005; pp. 437–461. [Google Scholar]
- Debelle, L.; Tamburro, A.M. Elastin: Molecular description and function. Int. J. Biochem. Cell Biol. 1999, 31, 261–272. [Google Scholar] [CrossRef]
- Yeo, G.C.; Aghaei-Ghareh-Bolagh, B.; Brackenreg, E.P.; Hiob, M.A.; Lee, P.; Weiss, A.S. Fabricated elastin. Adv. Healthc. Mater. 2015, 4, 2530–2556. [Google Scholar] [CrossRef] [PubMed]
- Urry, D.W. Molecular machines: How motion and other functions of living organisms can result from reversible chemical changes. Angew. Chem. Int. Ed. Engl. 1993, 32, 819–841. [Google Scholar] [CrossRef]
- Raju, K.; Anwar, R.A. Primary structures of bovine elastin a, b, and c deduced from the sequences of cDNA clones. J. Biol. Chem. 1987, 262, 5755–5762. [Google Scholar] [PubMed]
- Indik, Z.; Yeh, H.; Ornstein-Goldstein, N.; Sheppard, P.; Anderson, N.; Rosenbloom, J.C.; Peltonen, L.; Rosenbloom, J. Alternative splicing of human elastin mRNA indicated by sequence analysis of cloned genomic and complementary DNA. Proc. Natl. Acad. Sci. USA 1987, 84, 5680–5684. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.; Ornstein-Goldstein, N.; Indik, Z.; Sheppard, P.; Anderson, N.; Rosenbloom, J.C.; Cicila, G.; Yoon, K.; Rosenbloom, J. Sequence variation of bovine elastin mRNA due to alternative splicing. Coll. Relat. Res. 1987, 7, 235–247. [Google Scholar] [CrossRef]
- Sandberg, L.B.; Leslie, J.G.; Leach, C.T.; Alvarez, V.L.; Torres, A.R.; Smith, D.W. Elastin covalent structure as determined by solid phase amino acid sequencing. Pathol. Biol. 1985, 33, 266–274. [Google Scholar]
- Quiroz, F.G.; Chilkoti, A. Sequence heuristics to encode phase behaviour in intrinsically disordered protein polymers. Nat. Mater. 2015, 14, 1164–1171. [Google Scholar] [CrossRef] [Green Version]
- McDaniel, J.R.; Radford, D.C.; Chilkoti, A. A unified model for de novo design of elastin-like polypeptides with tunable inverse transition temperatures. Biomacromolecules 2013, 14, 2866–2872. [Google Scholar] [CrossRef]
- Wang, H.; Paul, A.; Nguyen, D.; Enejder, A.; Heilshorn, S.C. Tunable control of hydrogel microstructure by kinetic competition between self-assembly and crosslinking of elastin-like proteins. ACS Appl. Mater. Interfaces 2018, 10, 21808–21815. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.E.; Kong, G.A.; Dewhirst, M.W.; Zalutsky, M.R.; Chilkoti, A. Targeting a genetically engineered elastin-like polypeptide to solid tumors by local hyperthermia. Cancer Res. 2001, 61, 1548–1554. [Google Scholar] [PubMed]
- Reiersen, H.; Rees, A.R. Sodium sulphate reactivates a protein a minidomain with a short elastin β-Turn. Biochem. Biophys. Res. Commun. 2000, 276, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Cook, W.J.; Einspahr, H.; Trapane, T.L.; Urry, D.W.; Bugg, C.E. Crystal structure and conformation of the cyclic trimer of a repeat pentapeptide of elastin, cyclo-(l-valyl-l-prolylglycyl-l-valylglycyl)3. J. Am. Chem. Soc. 1980, 102, 5502–5505. [Google Scholar] [CrossRef]
- Khaled, M.A.; Venkatachalam, C.M.; Sugano, H.; Urry, D.W. Conformational characterization of cyclopentapeptide, lval-lpro-gly-lval-gly a repeating analogue of elastin. Int. J. Pept. Protein Res. 1981, 17, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.L.; Hong, M. Structure distribution in an elastin-mimetic peptide (VPGVG)3 investigated by solid-State NMR. J. Am. Chem. Soc. 2004, 126, 4199–4210. [Google Scholar] [CrossRef]
- Urry, D.W.; Mitchell, L.W.; Ohnishi, T. Studies on the conformation and interactions of elastin secondary structure of synthetic repeat hexapeptides. BBA-Protein Struct. 1975, 393, 296–306. [Google Scholar] [CrossRef]
- Urry, D.W.; Long, M.M.; Sugano, H. Cyclic analog of elastin polyhexapeptide exhibits an inverse temperature transition leading to crystallization. J. Biol. Chem. 1978, 253, 6301–6302. [Google Scholar]
- Schreiner, E.; Nicolini, C.; Ludolph, B.; Ravindra, R.; Otte, N.; Kohlmeyer, A.; Rousseau, R.; Winter, R.; Marx, D. Folding and unfolding of an elastinlike oligopeptide: “inverse temperature transition,” reentrance, and hydrogen-bond dynamics. Phys. Rev. Lett. 2004, 92, 148101. [Google Scholar] [CrossRef]
- Reiersen, H.; Clarke, A.R.; Rees, A.R. Short elastin-like peptides exhibit the same temperature-induced structural transitions as elastin polymers: Implications for protein engineering. J. Mol. Biol. 1998, 283, 255–264. [Google Scholar] [CrossRef]
- Pechar, M.; Brus, J.; Kostka, L.; Koňák, Č.; Urbanová, M.; Šlouf, M. Thermoresponsive self-assembly of short elastin-like polypentapeptides and their poly(ethylene glycol) derivatives. Macromol. Biosci. 2007, 7, 56–69. [Google Scholar] [CrossRef]
- Flamia, R.; Salvi, A.M.; D′Alessio, L.; Castle, J.E.; Tamburro, A.M. Transformation of amyloid-like fibers, formed from an elastin-based biopolymer, into a hydrogel: an X-ray photoelectron spectroscopy and atomic force microscopy study. Biomacromolecules 2006, 8, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Baer, M.; Schreiner, E.; Kohlmeyer, A.; Rousseau, R.; Marx, D. Inverse temperature transition of a biomimetic elastin model: reactive flux analysis of folding/unfolding and its coupling to solvent dielectric relaxation. J. Phys. Chem. B 2006, 110, 3576–3587. [Google Scholar] [CrossRef] [PubMed]
- Glaves, R.; Baer, M.; Schreiner, E.; Stoll, R.; Marx, D. Conformational dynamics of minimal elastin-like polypeptides: The role of proline revealed by molecular dynamics and nuclear magnetic resonance. ChemPhysChem 2008, 9, 2759–2765. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Menon, R.; Taite, L.J. Self-assembly of elastin-based peptides into the ECM: The importance of integrins and the elastin binding protein in elastic fiber assembly. Biomacromolecules 2011, 12, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Koga, T.; Nakamoto, K.; Odawara, K.; Matsuoka, T.; Higashi, N. Fabrication of thermo-responsive molecular layers from self-assembling elastin-like oligopeptides containing cell-binding domain for tissue engineering. Polymers 2015, 7, 134. [Google Scholar] [CrossRef]
- Taniguchi, S.; Watanabe, N.; Nose, T.; Maeda, I. Development of short and highly potent self-assembling elastin-derived pentapeptide repeats containing aromatic amino acid residues. J. Peptide Sci. 2016, 22, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Bochicchio, B.; Pepe, A.; Crudele, M.; Belloy, N.; Baud, S.; Dauchez, M. Tuning self-assembly in elastin-derived peptides. Soft Matter 2015, 11, 3385–3395. [Google Scholar] [CrossRef] [PubMed]
- Nuhn, H.; Klok, H.-A. Secondary structure formation and LCST behavior of short elastin-like peptides. Biomacromolecules 2008, 9, 2755–2763. [Google Scholar] [CrossRef]
- Iori, M.; Suguru, T.; Junko, E.; Noriko, W.; Takao, H.; Takeru, N. Comparison between coacervation property and secondary structure of synthetic peptides, Ile-containing elastin-derived pentapeptide repeats. Protein Peptide Lett. 2013, 20, 905–910. [Google Scholar]
- Suyama, K.; Tatsubo, D.; Iwasaki, W.; Miyazaki, M.; Kiyota, Y.; Takahashi, I.; Maeda, I.; Nose, T. Enhancement of self-aggregation properties of linear elastin-derived short peptides by simple cyclization: Strong self-aggregation properties of cyclo[FPGVG]n, consisting only of natural amino acids. Biomacromolecules 2018, 19, 3201–3211. [Google Scholar] [CrossRef] [PubMed]
- Scelsi, A.; Bochicchio, B.; Smith, A.; Saiani, A.; Pepe, A. Nanospheres from the self-assembly of an elastin-inspired triblock peptide. RSC Adv. 2015, 5, 95007–95013. [Google Scholar] [CrossRef]
- Wang, J.; Liu, K.; Xing, R.; Yan, X. Peptide self-assembly: Thermodynamics and kinetics. Chem. Soc. Rev. 2016, 45, 5589–5604. [Google Scholar] [CrossRef]
- Jeong, W.-J.; Kwon, S.H.; Lim, Y.-B. Modular self-assembling peptide platform with a tunable thermoresponsiveness via a single amino acid substitution. Adv. Funct. Mater. 2018, 28, 1803114. [Google Scholar] [CrossRef]
- Zhao, X.; Pan, F.; Xu, H.; Yaseen, M.; Shan, H.; Hauser, C.A.E.; Zhang, S.; Lu, J.R. Molecular self-assembly and applications of designer peptide amphiphiles. Chem. Soc. Rev. 2010, 39, 3480–3498. [Google Scholar] [CrossRef]
- Zhang, S. Fabrication of novel biomaterials through molecular self-assembly. Nat. Biotech. 2003, 21, 1171–1178. [Google Scholar] [CrossRef]
- Reches, M.; Gazit, E. Casting metal nanowires within discrete self-assembled peptide nanotubes. Science 2003, 300, 625–627. [Google Scholar] [CrossRef]
- Kim, T.G.; Shin, H.; Lim, D.W. Biomimetic scaffolds for tissue engineering. Adv. Funct. Mater. 2012, 22, 2446–2468. [Google Scholar] [CrossRef]
- Dou, X.-Q.; Feng, C.-L. Amino acids and peptide-based supramolecular hydrogels for three-dimensional cell culture. Adv. Mater. 2017, 29, 1604062. [Google Scholar] [CrossRef] [PubMed]
- Rad-Malekshahi, M.; Lempsink, L.; Amidi, M.; Hennink, W.E.; Mastrobattista, E. Biomedical applications of self-assembling peptides. Bioconjugate Chem. 2016, 27, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Wang, Y.; Zhao, W.; Qi, R.; Han, Y.; Wu, R.; Wang, Y.; Xu, H. Peptide-induced DNA condensation into virus-mimicking nanostructures. ACS Appl. Mater. Interfaces 2018, 10, 24349–24360. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Lu, S.; Zhao, W.; Deng, L.; Wang, M.; Wang, J.; Zhou, P.; Wang, D.; Xu, H.; Lu, J.R. Peptide self-assembled nanostructures with distinct morphologies and properties fabricated by molecular design. ACS Appl. Mater. Interfaces 2017, 9, 39174–39184. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Pritzker, M.; Fung, S.Y.; Sheng, Y.; Wang, W.; Chen, P. Anion effect on the nanostructure of a metal ion binding self-assembling peptide. Langmuir 2006, 22, 8553–8562. [Google Scholar] [CrossRef] [PubMed]
- Mart, R.J.; Osborne, R.D.; Stevens, M.M.; Ulijn, R.V. Peptide-based stimuli-responsive biomaterials. Soft Matter 2006, 2, 822–835. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Y.; Ge, X.; Han, S.; Wang, S.; Zhou, P.; Shan, H.; Zhao, X.; Lu, J.R. Twisted nanotubes formed from ultrashort amphiphilic peptide I3K and their templating for the fabrication of silica nanotubes. Chem.Mater. 2010, 22, 5165–5173. [Google Scholar] [CrossRef]
- Wang, S.; Ge, X.; Xue, J.; Fan, H.; Mu, L.; Li, Y.; Xu, H.; Lu, J.R. Mechanistic processes underlying biomimetic synthesis of silica nanotubes from self-assembled ultrashort peptide templates. Chem. Mater. 2011, 23, 2466–2474. [Google Scholar] [CrossRef]
- Castelletto, V.; Hamley, I.W. Self assembly of a model amphiphilic phenylalanine peptide/polyethylene glycol block copolymer in aqueous solution. Biophys. Chem. 2009, 141, 169–174. [Google Scholar] [CrossRef]
- Cao, M.; Cao, C.; Zhang, L.; Xia, D.; Xu, H. Tuning of peptide assembly through force balance adjustment. J. Colloid Interface Sci. 2013, 407, 287–295. [Google Scholar] [CrossRef]
- Aguiar, J.; Carpena, P.; Molina-Bolívar, J.A.; Carnero Ruiz, C. On the determination of the critical micelle concentration by the pyrene 1:3 ratio method. J. Colloid Interface Sci. 2003, 258, 116–122. [Google Scholar] [CrossRef]
- Arad, O.; Goodman, M. Depsipeptide analogues of elastin repeating sequences: Conformational analysis. Biopolymers 1990, 29, 1651–1668. [Google Scholar] [CrossRef]
- Paramonov, S.E.; Jun, H.W.; Hartgerink, J.D. Self-assembly of peptide-amphiphile nanofibers: The roles of hydrogen bonding and amphiphilic packing. J. Am. Chem. Soc. 2006, 128, 7291–7298. [Google Scholar] [CrossRef] [PubMed]
- Pelton, J.T.; McLean, L.R. Spectroscopic methods for analysis of protein secondary structure. Anal. Biochem. 2000, 277, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Cao, C.; Zhou, P.; Wang, N.; Wang, D.; Wang, J.; Xia, D.; Xu, H. Self-assembly of amphiphilic peptides: Effects of the single-chain-to-gemini structural transition and the side chain groups. Colloids Surf. A 2015, 469, 263–270. [Google Scholar] [CrossRef]
- Mazor, Y.; Gilead, S.; Benhar, I.; Gazit, E. Identification and characterization of a novel molecular-recognition and self-assembly domain within the Islet amyloid polypeptide. J. Mol. Biol. 2002, 322, 1013–1024. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Sequence | Abbreviation |
|---|---|
| Ac-KGVPGVGK-NH2 | K-K8 |
| Ac-GVPGVGKK-NH2 | KK8 |
| Ac-IIIKGVPGVGK-NH2 | IK-K11 |
| Ac-IIIGVPGVGKK-NH2 | IKK11 |
| Ac-IIIKGVPGVGVPGVGK-NH2 | IK-K16 |
| Ac-IIIGVPGVGVPGVGKK-NH2 | IKK16 |
| Peptide | Temperature | CAC (mM) | Self-Assembled Structures | Size Parameters (nm) | |
|---|---|---|---|---|---|
| Length | Diameter | ||||
| K-K8 | 20 °C | >8.0 | amorphous aggregates | — | — |
| 80 °C | — | amorphous aggregates | — | — | |
| KK8 | 20 °C | >8.0 | amorphous aggregates | — | — |
| 80 °C | — | amorphous aggregates | — | — | |
| IK-K11 | 20 °C | 2.65 ± 0.26 | short fibrils | <600 | 11.0 ± 2.5 |
| 80 °C | — | long smooth fibrils | >1000 | 15.0 ± 8.0 | |
| IKK11 | 20 °C | 2.20 ± 0.17 | short fibrils | <1200 | 20.0 ± 3.5 |
| 80 °C | — | long smooth fibrils | >1000 | 12.0 ± 2.0 | |
| IK-K16 | 20 °C | 0.94 ± 0.11 | long smooth fibrils | >1000 | 13.5 ± 2.5 |
| 80 °C | — | long smooth fibrils & fibril bundles | >1000 | 14.0 ± 2.0 | |
| IKK16 | 20 °C | 0.74 ± 0.08 | long rough fibrils | >1000 | 20.0 ± 5.5 |
| 80 °C | — | long smooth fibrils | >2000 | 11.0 ± 1.0 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, M.; Shen, Y.; Wang, Y.; Wang, X.; Li, D. Self-Assembly of Short Elastin-like Amphiphilic Peptides: Effects of Temperature, Molecular Hydrophobicity and Charge Distribution. Molecules 2019, 24, 202. https://doi.org/10.3390/molecules24010202
Cao M, Shen Y, Wang Y, Wang X, Li D. Self-Assembly of Short Elastin-like Amphiphilic Peptides: Effects of Temperature, Molecular Hydrophobicity and Charge Distribution. Molecules. 2019; 24(1):202. https://doi.org/10.3390/molecules24010202
Chicago/Turabian StyleCao, Meiwen, Yang Shen, Yu Wang, Xiaoling Wang, and Dongxiang Li. 2019. "Self-Assembly of Short Elastin-like Amphiphilic Peptides: Effects of Temperature, Molecular Hydrophobicity and Charge Distribution" Molecules 24, no. 1: 202. https://doi.org/10.3390/molecules24010202