Genome-Wide Identification and Comparative Analysis for OPT Family Genes in Panax ginseng and Eleven Flowering Plants

Abstract
:1. Introduction
2. Results and Discussion
2.1. Identification of OPT Genes in P. ginseng and 11 Other Flowering Plants
2.2. Protein Properties of OPT Genes for OPT Genes Identified in P. ginseng and 11 Other Flowering Plants
2.3. Phylogenetic Analyses, Classification, and Functional Relatedness of the OPT Genes Identified in P. ginseng and 11 Other Flowering Plants
2.4. Conserved Domains and Motif Analysis for OPT Genes Identified in P. ginseng and 11 Other Flowering Plants
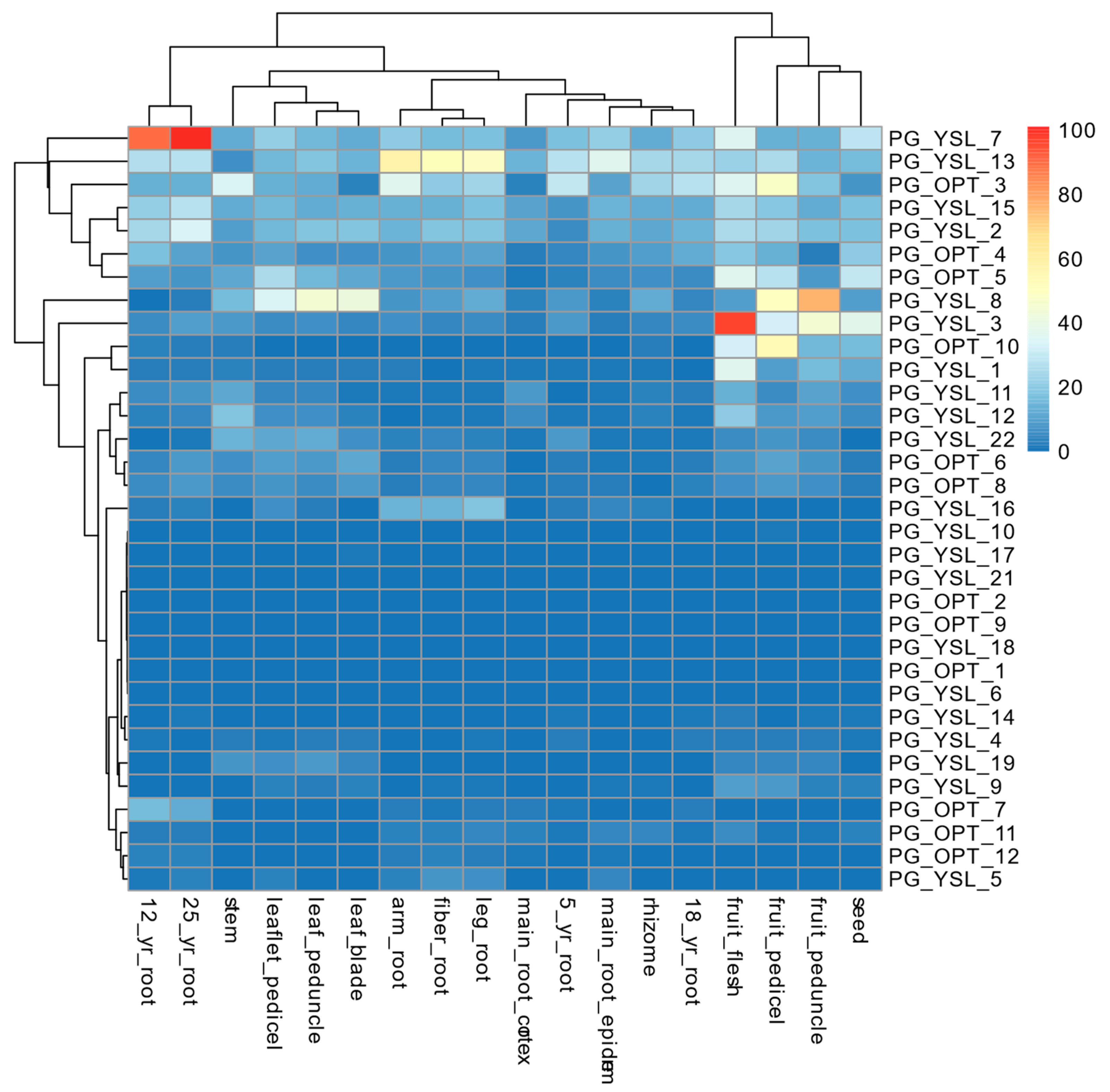
2.5. Profiling of Expression Patterns for OPT Genes Identified in P. ginseng
2.6. Analysis of Co-Expression Network between OPT Genes and Potent Transcription factOr for P. ginseng
3. Materials and Methods
3.1. Sequence Retrieval and Identification of OPT Genes
3.2. Phylogenetic Analysis for OPT Genes
3.3. Estimation of Duplication Time for OPT Paralogs
3.4. Analysis of Motif Composition for OPT Genes
3.5. Profiling Expression of OPT Genes for P. ginseng
3.6. Identification of Regulatory Network between OPT Genes and Transcription Factors for P. ginseng
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Stacey, M.G.; Koh, S.; Becker, J.; Stacey, G. Atopt3, a member of the oligopeptide transporter family, is essential for embryo development in Arabidopsis. Plant Cell 2002, 14, 2799–2811. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.R.; Basrai, M.A.; Steiner, H.Y.; Naider, F.; Becker, J.M. Isolation and characterization of a saccharomyces cerevisiae peptide transport gene. Mol. Cell. Biol. 1994, 14, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Steiner, H.Y.; Naider, F.; Becker, J.M. The ptr family: A new group of peptide transporters. Mol. Microbiol. 1995, 16, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Swift, S.; Throup, J.P.; Williams, P.; Salmond, G.P.; Stewart, G.S. Quorum sensing: A population-density component in the determination of bacterial phenotype. Trends Biochem. Sci. 1996, 21, 214–219. [Google Scholar] [CrossRef]
- Kuchler, K.; Sterne, R.E.; Thorner, J. Saccharomyces cerevisiae ste6 gene product: A novel pathway for protein export in eukaryotic cells. Embo J. 1989, 8, 3973–3984. [Google Scholar] [CrossRef] [PubMed]
- Curie, C.; Panaviene, Z.; Loulergue, C.; Dellaporta, S.L.; Briat, J.-F.; Walker, E.L. Maize yellow stripe1 encodes a membrane protein directly involved in Fe(III) uptake. Nature 2001, 409, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Stacey, M.G.; Patel, A.; Mcclain, W.E.; Mathieu, M.; Remley, M.; Rogers, E.E.; Gassmann, W.; Blevins, D.G.; Stacey, G. The Arabidopsis atopt3 protein functions in metal homeostasis and movement of iron to developing seeds. Plant Physiol. 2008, 146, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Hamon, Y.; Chimini, G. The human atp-binding cassette (abc) transporter superfamily. J. Lipid Res. 2001, 42, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F. Abc transporters: From microorganisms to man. Ann. Rev. Cell Biol. 1992, 8, 67–113. [Google Scholar] [CrossRef]
- Hauser, M.; Narita, V.; Donhardt, A.M.; Naider, F.; Becker, J.M. Multiplicity and regulation of genes encoding peptide transporters in saccharomyces cerevisiae. Mol. Membr. Biol. 2001, 18, 105–112. [Google Scholar] [CrossRef]
- Newstead, S. Recent advances in understanding proton coupled peptide transport via the pot family. Curr. Opin. Struct. Biol. 2017, 45, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.; Miller, A. Transporters responsible for the uptake and partitioning of nitrogenous solutes. Ann. Rev. Plant Biol. 2001, 52, 659–688. [Google Scholar] [CrossRef] [PubMed]
- Lubkowitz, M. The opt family functions in long-distance peptide and metal transport in plants. In Genetic Engineering: Principles and Methods; Setlow, J.K., Ed.; Springer: Boston, MA, USA, 2006; pp. 35–55. [Google Scholar]
- Lubkowitz, M.A.; Hauser, L.; Breslav, M.; Naider, F.; Becker, J.M. An oligopeptide transport gene from candida albicans. Microbiology 1997, 143, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Lubkowitz, M.A.; Barnes, D.; Breslav, M.; Burchfield, A.; Naider, F.; Becker, J.M. Schizosaccharomyces pombe isp4 encodes a transporter representing a novel family of oligopeptide transporters. Mol. Microbiol. 1998, 28, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Lubkowitz, M. The oligopeptide transporters: A small gene family with a diverse group of substrates and functions? Mol. Plant 2011, 4, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Gomolplitinant, K.M.; Saier, M., Jr. Evolution of the oligopeptide transporter family. J. Membr. Biol. 2011, 240, 89. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Tan, J.; Zhang, Y.; Liang, S.; Xiang, S.; Wang, H.; Chai, T. Isolation and characterization of a novel cadmium-regulated yellow stripe-like transporter (snysl3) in solanum nigrum. Plant Cell Rep. 2017, 36, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Murata, Y.; Ma, J.F.; Yamaji, N.; Ueno, D.; Nomoto, K.; Iwashita, T. A specific transporter for iron(III)-phytosiderophore in barley roots. Plant J. 2006, 46, 563–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, S.; Wiles, A.M.; Sharp, J.S.; Naider, F.R.; Becker, J.M.; Stacey, G. An oligopeptide transporter gene family in arabidopsis. Plant Physiol. 2002, 128, 21–29. [Google Scholar] [CrossRef]
- Wongkaew, A.; Asayama, K.; Kitaiwa, T.; Nakamura, S.-I.; Kojima, K.; Stacey, G.; Sekimoto, H.; Yokoyama, T.; Ohkama-Ohtsu, N. Atopt6 protein functions in long-distance transport of glutathione in arabidopsis thaliana. Plant Cell Physiol. 2018. [Google Scholar] [CrossRef]
- Zhang, Z.; Xie, Q.; Jobe, T.O.; Kau, A.R.; Wang, C.; Li, Y.; Qiu, B.; Wang, Q.; Mendoza-Cózatl, D.G.; Schroeder, J.I. Identification of atopt4 as a plant glutathione transporter. Mol. Plant 2016, 9, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Cózatl, D.G.; Xie, Q.; Akmakjian, G.Z.; Jobe, T.O.; Patel, A.; Stacey, M.G.; Song, L.; Demoin, D.W.; Jurisson, S.S.; Stacey, G. Opt3 is a component of the iron-signaling network between leaves and roots and misregulation of opt3 leads to an over-accumulation of cadmium in seeds. Mol. Plant 2014, 7, 1455–1469. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, M.W.; Li, G.W.; Lubkowitz, M.A.; Grusak, M.A. Characterization of the pt clade of oligopeptide transporters in rice. Plant Genome 2008, 1, 77–88. [Google Scholar] [CrossRef]
- Bogs, J.; Bourbouloux, A.; Cagnac, O.; Wachter, A.; Rausch, T.; Delrot, S. Functional characterization and expression analysis of a glutathione transporter, bjgt1, from brassica juncea: Evidence for regulation by heavy metal exposure. Plant Cell Environ. 2003, 26, 1703–1711. [Google Scholar] [CrossRef]
- Carole, D.M.; Beno, T.P. Role of glutathione in plant signaling under biotic stress. Plant Signal. Behav. 2012, 7, 210–212. [Google Scholar] [Green Version]
- Yen, M.-R.; Tseng, Y.-H.; Saie, M., Jr. Maize yellow stripe1, an iron-phytosiderophore uptake transporter, is a member of the oligopeptide transporter (opt) family. Microbiology 2001, 147, 2881–2883. [Google Scholar] [CrossRef] [PubMed]
- Cobbett, C.; Goldsbrough, P. Phytochelatins and metallothioneins: Roles in heavy metal detoxification and homeostasis. Annu. Rev. Plant Biol. 2003, 53, 159–182. [Google Scholar] [CrossRef]
- Bourbouloux, A.; Shahi, P.; Chakladar, A.; Delrot, S.; Bachhawat, A.K. Hgt1p, a high affinity glutathione transporter from the yeast saccharomyces cerevisiae. J. Biol. Chem. 2000, 275, 13259–13265. [Google Scholar] [CrossRef]
- Liu, T.; Zeng, J.; Xia, K.; Fan, T.; Li, Y.; Wang, Y.; Xu, X.; Zhang, M. Evolutionary expansion and functional diversification of oligopeptide transporter gene family in rice. Rice 2012, 5, 1–14. [Google Scholar] [CrossRef]
- Xiang, Q.; Shen, K.; Yu, X.; Zhao, K.; Gu, Y.; Zhang, X.; Chen, X.; Chen, Q. Analysis of the oligopeptide transporter gene family in ganoderma lucidum: Structure, phylogeny, and expression patterns. Genome 2017, 60, 293–302. [Google Scholar] [CrossRef]
- Cao, J.; Huang, J.; Yang, Y.; Hu, X. Analyses of the oligopeptide transporter gene family in poplar and grape. BMC Genom. 2011, 12, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, T.K. Brief introduction of panax ginseng c.A. Meyer. J. Korean Med. Sci. 2001, 16 (Suppl.), S3–S5. [Google Scholar]
- Jiang, X.; Yang, C.; Baosheng, L.; Shuiming, X.; Qinggang, Y.; Rui, B.; He, S.; Linlin, D.; Xiwen, L.; Jun, Q. Panax ginseng genome examination for ginsenoside biosynthesis. GigaScience 2017, 6, 1–15. [Google Scholar]
- Li, H.; Benedito, V.A.; Udvardi, M.K.; Zhao, P.X. Transporttp: A two-phase classification approach for membrane transporter prediction and characterization. BMC Bioinform. 2009, 10, 418. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Shintaro, K.; Haruhiko, I.; Daichi, M.; Michiko, T.; Hiromi, N.; Satoshi, M.; Nishizawa, N.K. Osysl2 is a rice metal-nicotianamine transporter that is regulated by iron and expressed in the phloem. Plant J. 2010, 39, 415–424. [Google Scholar]
- Haruhiko, I.; Takanori, K.; Tomoko, N.; Michiko, T.; Yusuke, K.; Kazumasa, S.; Mikio, N.; Hiromi, N.; Satoshi, M.; Nishizawa, N.K. Rice osysl15 is an iron-regulated iron(III)-deoxymugineic acid transporter expressed in the roots and is essential for iron uptake in early growth of the seedlings. J. Biol. Chem. 2009, 284, 3470–3479. [Google Scholar]
- DiDonido, D., Jr.; Roberts, L.A.; Sanderson, T.; Eisley, R.B.; Walker, E.L. Arabidopsis yellow stripe-like2 (ysl2): A metal-regulated gene encoding a plasma membrane transporter of nicotianamine–metal complexes. Plant J. 2004, 39, 403–414. [Google Scholar] [CrossRef]
- Waters, B.M.; Chu, H.H.; Didonato, R.J.; Roberts, L.A.; Eisley, R.B.; Lahner, B.; Salt, D.E.; Walker, E.L. Mutations in arabidopsis yellow stripe-like1 and yellow stripe-like3 reveal their roles in metal ion homeostasis and loading of metal ions in seeds. Plant Physiol. 2006, 141, 1446–1458. [Google Scholar] [CrossRef]
- Marie, L.J.; Adam, S.; Stéphane, M.; Jean-François, B.; Catherine, C. A loss-of-function mutation in atysl1 reveals its role in iron and nicotianamine seed loading. Plant J. 2010, 44, 769–782. [Google Scholar]
- Cagnac, O.; Bourbouloux, A.; Chakrabarty, D.; Zhang, M.-Y.; Delrot, S. Atopt6 transports glutathione derivatives and is induced by primisulfuron. Plant Physiol. 2004, 135, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Pike, S.; Patel, A.; Stacey, G.; Gassmann, W. Arabidopsis opt6 is an oligopeptide transporter with exceptionally broad substrate specificity. Plant Cell Physiol. 2009, 50, 1923–1932. [Google Scholar] [CrossRef] [PubMed]
- Jaillon, O.; Aury, J.M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar] [PubMed] [Green Version]
- Marchlerbauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R. Cdd/sparcle: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Su, H.; Zhang, L.; Liao, B.S.; Xiao, S.M.; Dong, L.L.; Hu, Z.G.; Wang, P.; Li, X.W.; Huang, Z.H. Comprehensive characterization for ginsenosides biosynthesis in ginseng root by integration analysis of chemical and transcriptome. Molecules 2017, 22, 889. [Google Scholar] [CrossRef]
- Wang, K.; Jiang, S.; Sun, C.; Lin, Y.; Rui, Y.; Yi, W.; Zhang, M. The spatial and temporal transcriptomic landscapes of ginseng, panax ginseng c. A. Meyer. Sci. Rep. 2015, 5, 18283. [Google Scholar] [CrossRef]
- Osawa, H.; Stacey, G.; Gassmann, W. Scopt1 and atopt4 function as proton-coupled oligopeptide transporters with broad but distinct substrate specificities. Biochem. J. 2006, 393, 267–275. [Google Scholar] [CrossRef]
- Prince, V.E.; Pickett, F.B. Splitting pairs: The diverging fates of duplicated genes. Nat. Rev. Genet. 2002, 3, 827–837. [Google Scholar] [CrossRef]
- Dai, X.; Sinharoy, S.; Udvardi, M.; Zhao, P.X. Planttfcat: An online plant transcription factor and transcriptional regulator categorization and analysis tool. BMC Bioinform. 2013, 14, 321. [Google Scholar] [CrossRef]
- Sapna, K.; Nie, J.; Chen, H.S.; Hao, M.; Ron, S.; Xiang, L.; Lu, M.Z.; Taylor, W.M.; Wei, H. Evaluation of gene association methods for coexpression network construction and biological knowledge discovery. PLoS ONE 2012, 7, e50411. [Google Scholar]
- Goodstein, D.M.; Shu, S.; Russell, H.; Rochak, N.; Hayes, R.D.; Joni, F.; Therese, M.; William, D.; Uffe, H.; Nicholas, P. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.Y.; Wagner, J.R.; Laird, M.R.; Melli, G.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J.; Brinkman, F.S.L. Psortb 3.0. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Horton, P.; Park, K.J.; Obayashi, T.; Fujita, N.; Harada, H.; Adamscollier, C.J.; Nakai, K. Wolf psort: Protein localization predictor. Nucleic Acids Res. 2007, 35, 585–587. [Google Scholar] [CrossRef] [PubMed]
- Marchlerbauer, A.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; Deweesescott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; Gwadz, M. Cdd: Specific functional annotation with the conserved domain database. Nucleic Acids Res. 2009, 37, D205–D210. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Lu, S.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; et al. Cdd: A conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 2011, 39, D225–D229. [Google Scholar] [CrossRef]
- Katoh, K.; Kuma, K.; Toh, H.; Miyata, T. Mafft version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef]
- Schliep, K.P. Phangorn: Phylogenetic analysis in r. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef]
- Stamatakis, A. Raxml version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (Itol): An Online Tool for Phylogenetic Tree Display and Annotation; Oxford University Press: Oxford, UK, 2007; pp. 78–82. [Google Scholar]
- Suyama, M.; Torrents, D.; Bork, P. Pal2nal: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef]
- Yang, Z. Paml: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. Cabios 1997, 13, 555–556. [Google Scholar] [CrossRef] [PubMed]
- The Potato Genome Sequencing Consortium; Xu, X.; Pan, S.; Cheng, S.; Zhang, B.; Mu, D.; Ni, P.; Zhang, G.; Yang, S.; Li, R.; et al. Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, N.D.; Debellé, F.; Oldroyd, G.E.D.; Geurts, R.; Cannon, S.B.; Udvardi, M.K.; Benedito, V.A.; Mayer, K.F.X.; Gouzy, J.; Schoof, H.; et al. The medicago genome provides insight into the evolution of rhizobial symbioses. Nature 2011, 480, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S. Ncbi sra toolkit technology for next generation sequence data. Pump Ind. Anal. 2000, 3, 2230–2234. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of rna-seq experiments with hisat, stringtie and ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Kolde, R. Pheatmap: Pretty Heatmaps. R Package Version 1.0.8. Available online: https://CRAN.R-project.org/package=pheatmap (accessed on 18 December 2018).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Root samples of the Panax ginseng are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Predicted | De-Redundant | Final |
|---|---|---|---|
| Ginseng | 39 | 37 | 37 |
| Arabidopsis | 18 | 16 | 17 |
| Rice | 12 | 10 | 18 |
| Sorghum | 38 | 26 | 26 |
| Carrot | 25 | 16 | 16 |
| Potato | 42 | 29 | 29 |
| Tomato | 23 | 17 | 17 |
| Cassava | 29 | 21 | 21 |
| Clover | 31 | 25 | 25 |
| Cacao | 27 | 19 | 19 |
| Poplar | 54 | 28 | 28 |
| Grape | 26 | 24 | 23 |
| Total | 364 | 268 | 276 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, H.; Chu, Y.; Bai, J.; Gong, L.; Huang, J.; Xu, W.; Zhang, J.; Qiu, X.; Xu, J.; Huang, Z. Genome-Wide Identification and Comparative Analysis for OPT Family Genes in Panax ginseng and Eleven Flowering Plants. Molecules 2019, 24, 15. https://doi.org/10.3390/molecules24010015
Su H, Chu Y, Bai J, Gong L, Huang J, Xu W, Zhang J, Qiu X, Xu J, Huang Z. Genome-Wide Identification and Comparative Analysis for OPT Family Genes in Panax ginseng and Eleven Flowering Plants. Molecules. 2019; 24(1):15. https://doi.org/10.3390/molecules24010015
Chicago/Turabian StyleSu, He, Yang Chu, Junqi Bai, Lu Gong, Juan Huang, Wen Xu, Jing Zhang, Xiaohui Qiu, Jiang Xu, and Zhihai Huang. 2019. "Genome-Wide Identification and Comparative Analysis for OPT Family Genes in Panax ginseng and Eleven Flowering Plants" Molecules 24, no. 1: 15. https://doi.org/10.3390/molecules24010015