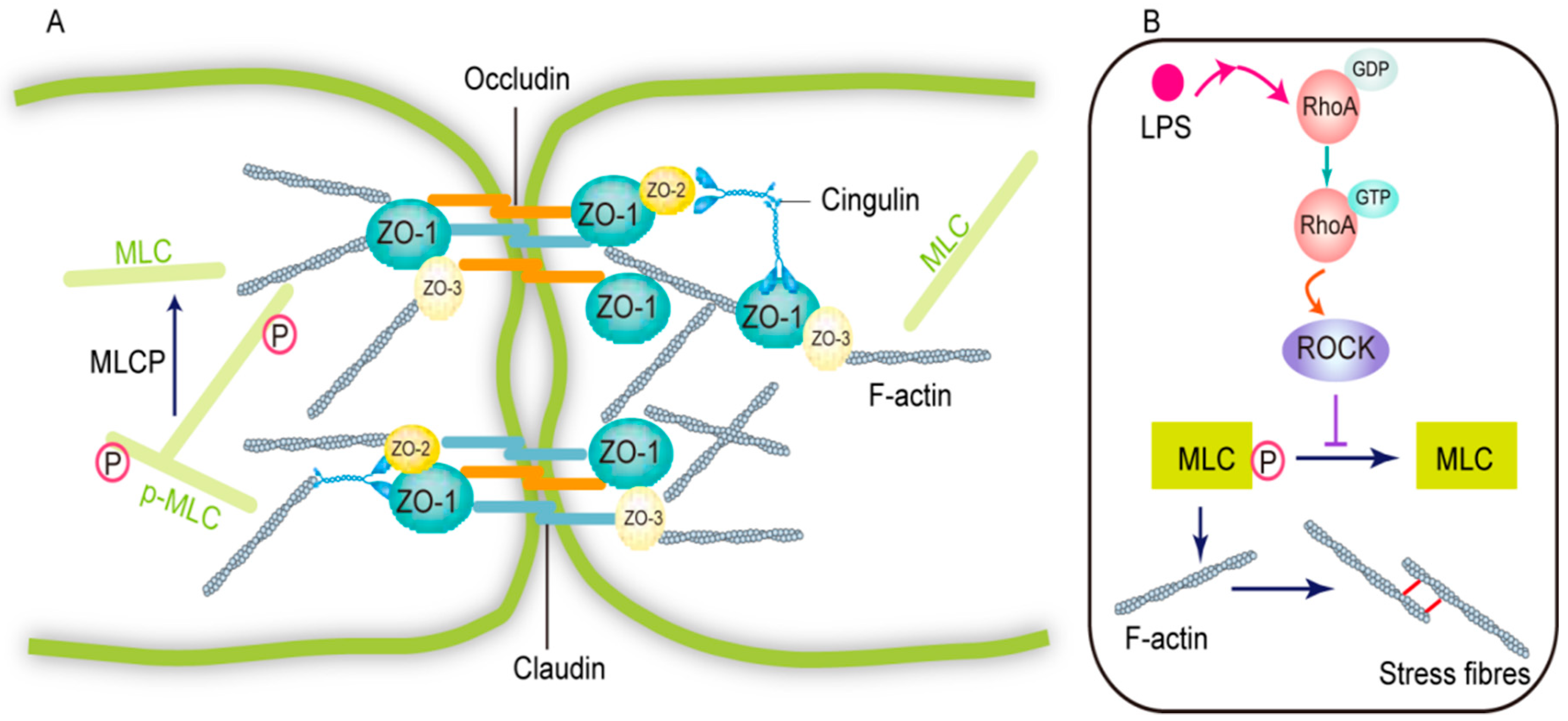
RhoA/ROCK-2 Pathway Inhibition and Tight Junction Protein Upregulation by Catalpol Suppresses Lipopolysaccaride-Induced Disruption of Blood-Brain Barrier Permeability


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Antibodies
2.2. Isolation and Purification of Rat BMECs
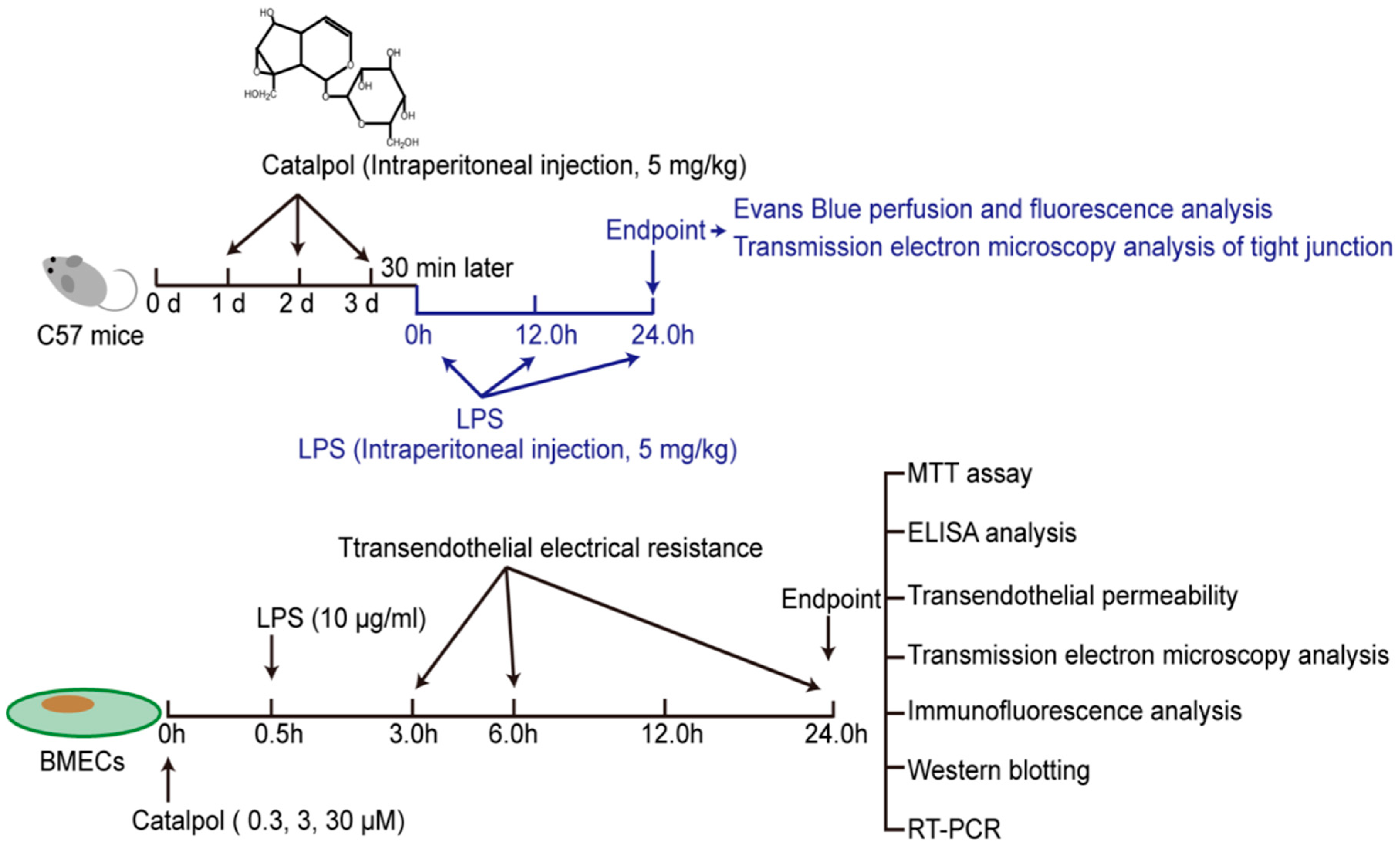
2.3. In Vivo LPS Challenge and Catalpol Protective Effect
2.4. In Vitro LPS Challenge and Catalpol Protective Effect
2.4.1. MTT Assay for Cell Viability
2.4.2. Measurement of Transendothelial Electrical Resistance (TEER)
2.4.3. Determination of the Transendothelial Permeability by Sodium Fluorescein
2.4.4. Immunofluorescence Analysis
2.4.5. ELISA Analysis
2.4.6. Transmission Electron Microscopy Analysis of Tight Junction
2.4.7. RT-PCR and qPCR
2.4.8. Western Blot Analysis
2.5. Statistics
3. Results
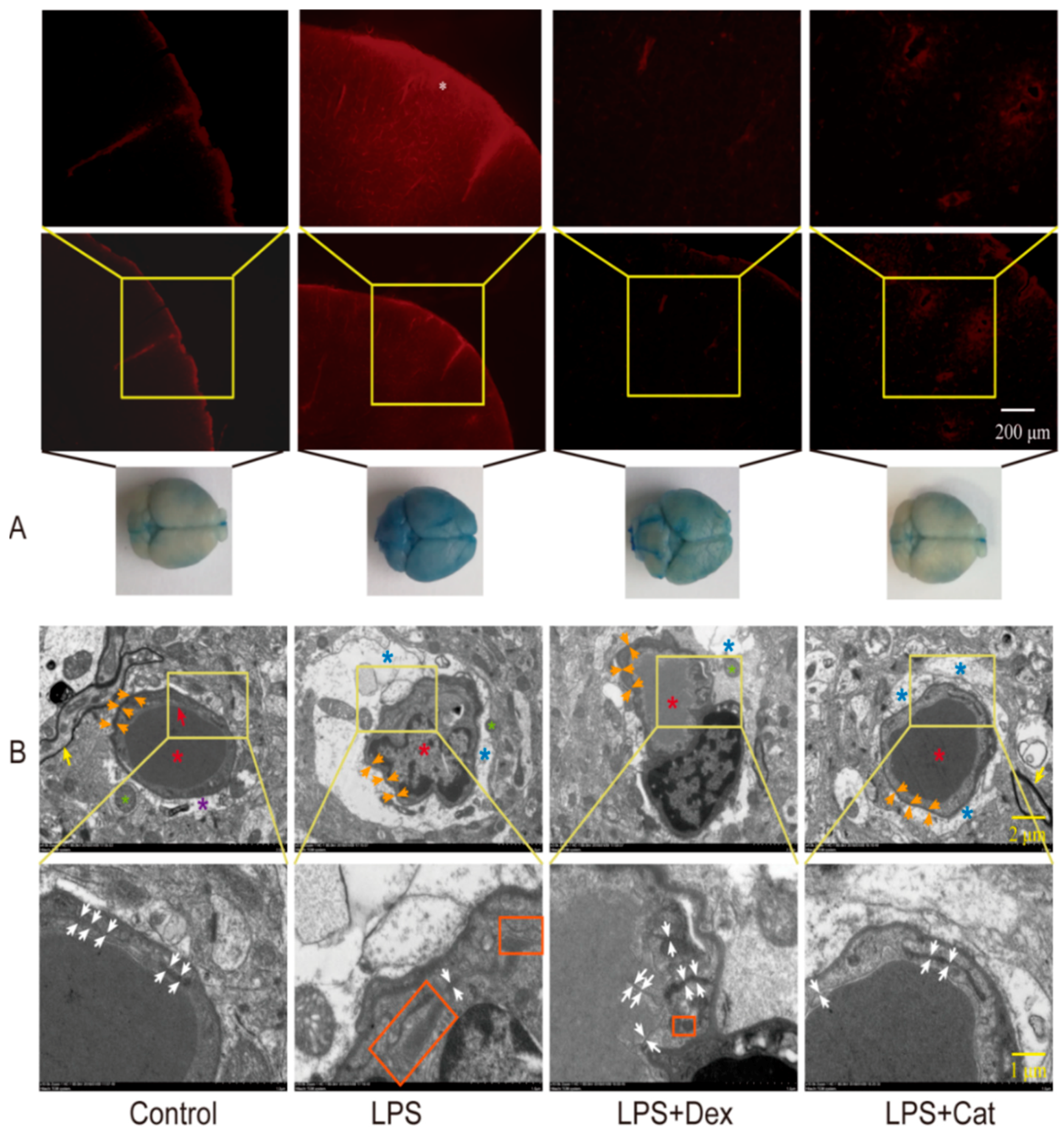
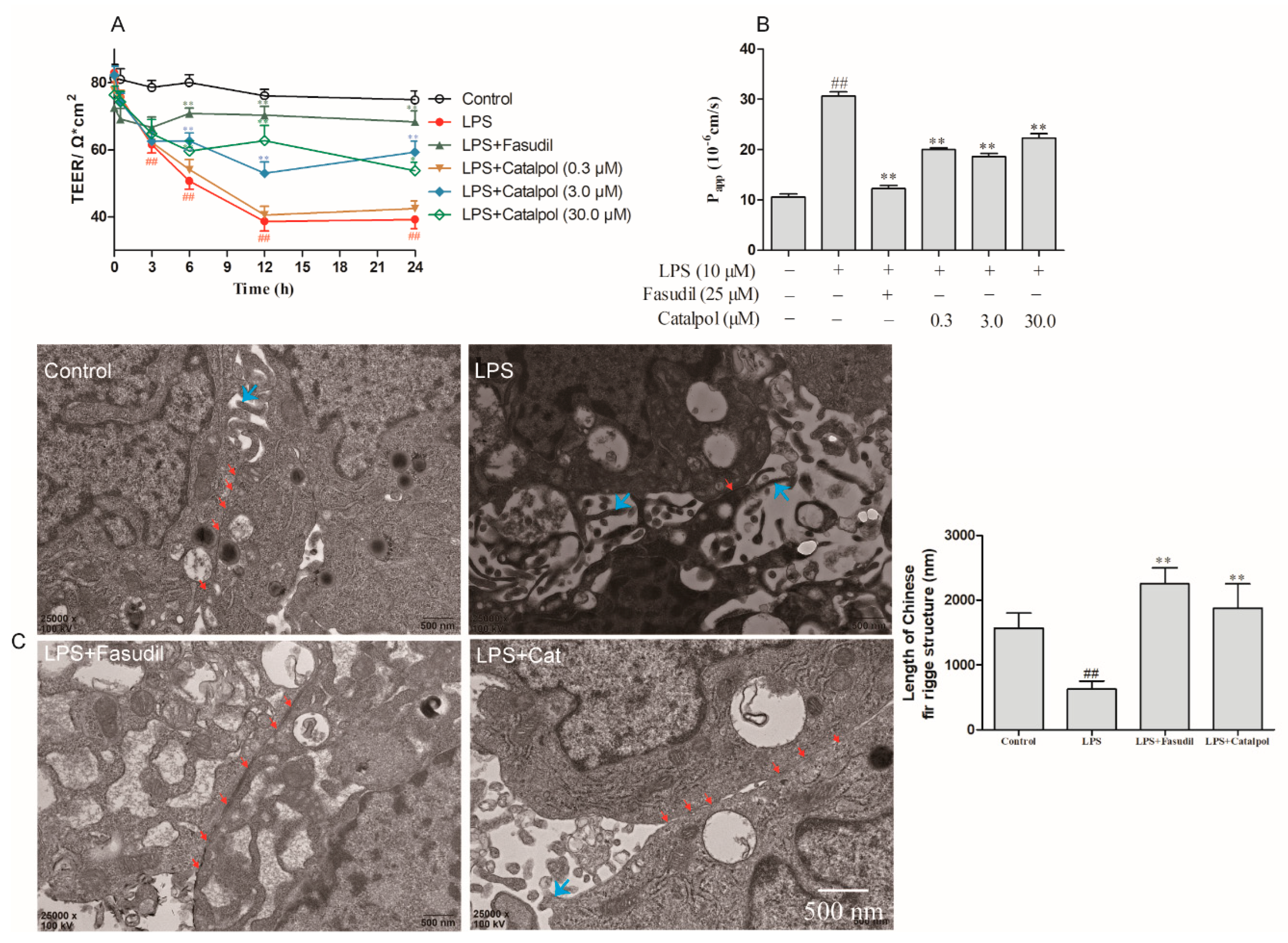
3.1. Catalpol Alleviated the Reduction in LPS-Induced BBB Permeability
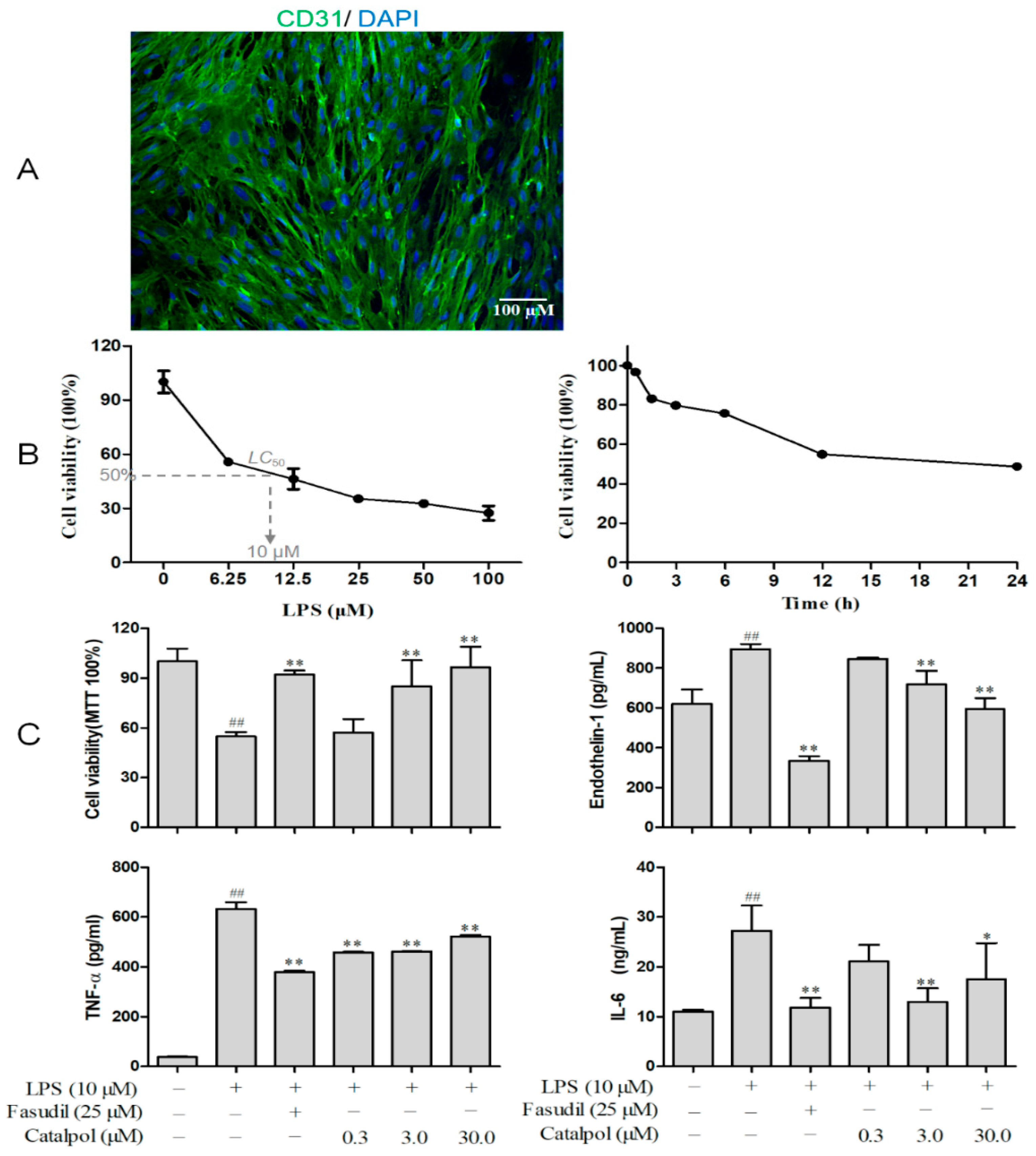
3.2. BMEC Characterization and Screening for LPS Concentration and Treatment Time
3.3. Catalpol Improved BMEC Survival and Its Secretory Function
3.4. Catalpol Alleviated the Reduction in LPS-Induced BMEC Permeability
3.5. Catalpol Antagonized the Changes in the Ultrastructure of BMEC Tight Junction Caused by LPS Trauma
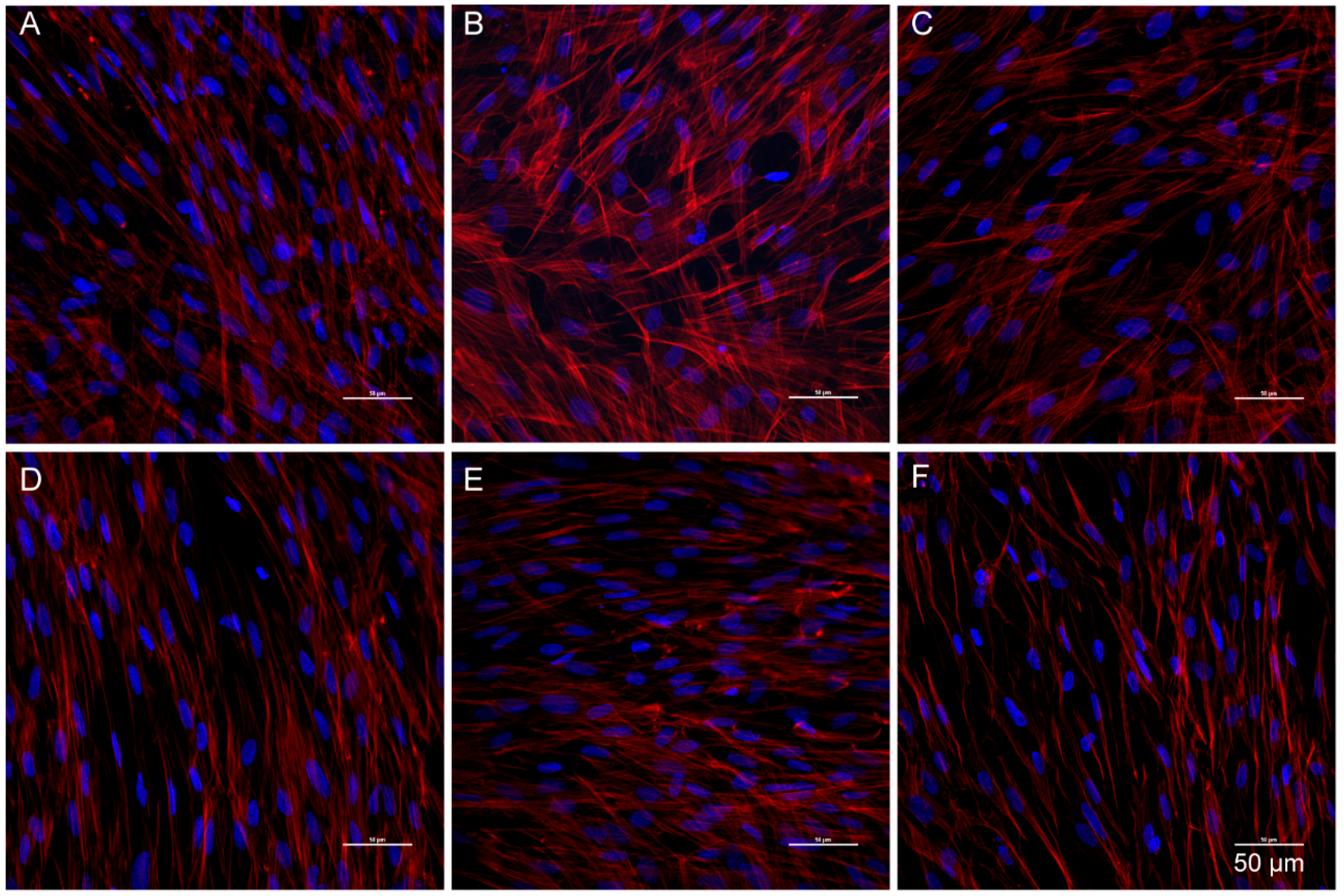
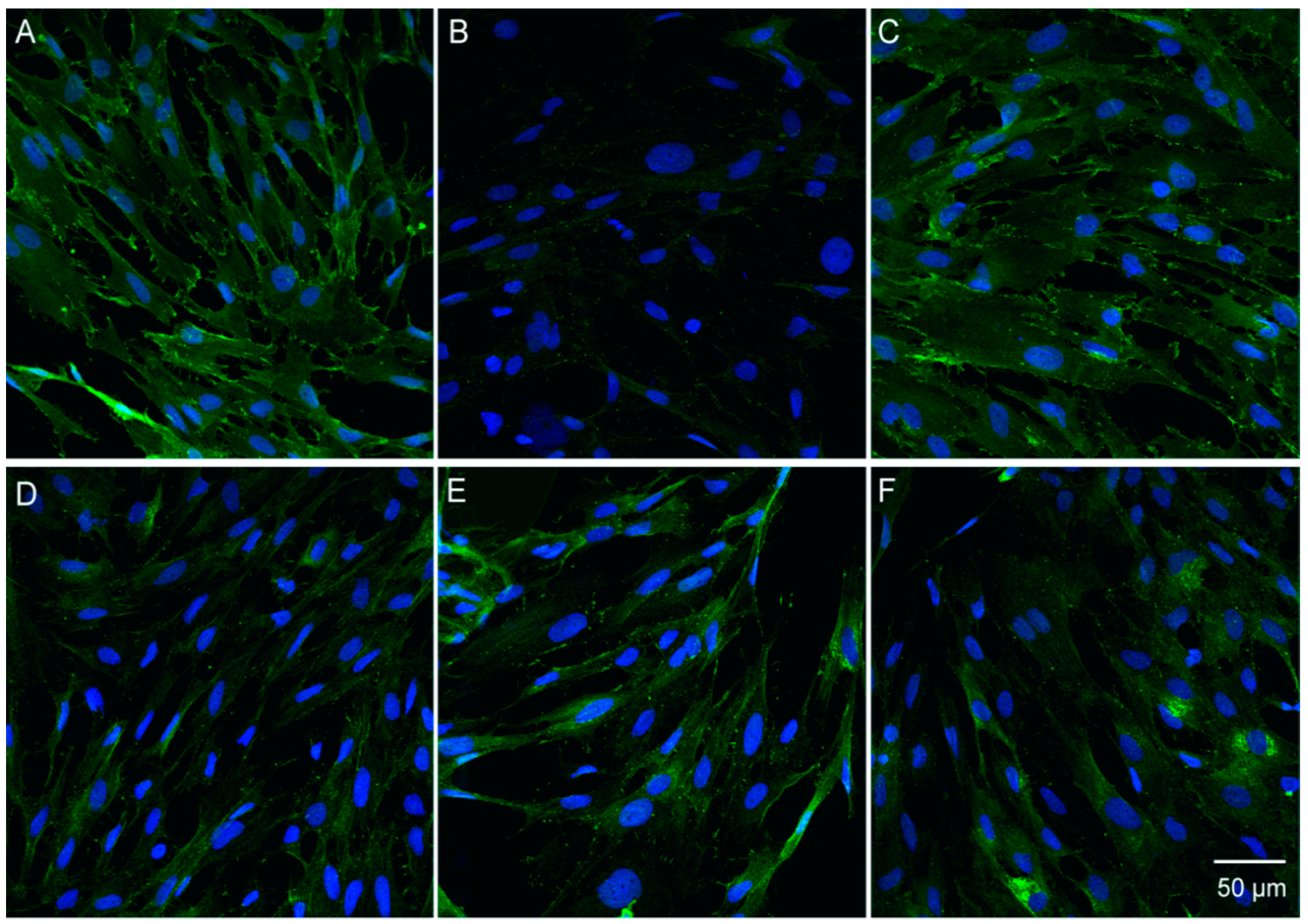
3.6. Catalpol Antagonized F-Actin Disaggregation and Downregulation of the LPS-Induced Expression of ZO-1 and Claudin-5
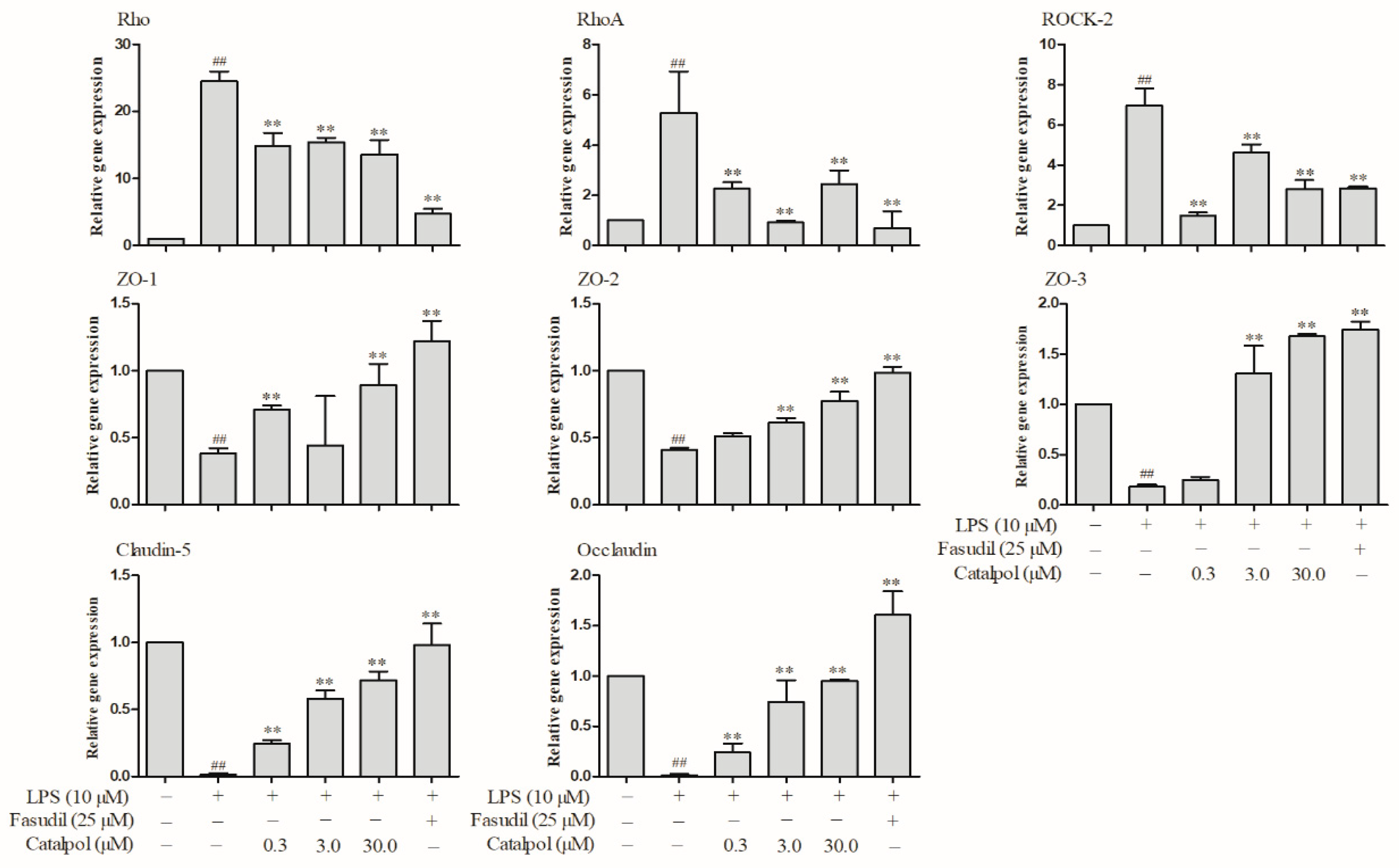
3.7. Catalpol Antagonized Changes in the Protein Expressions Relevant to BMEC Tight Junction and RhoA/ROCK Signaling Pathway Caused by LPS Trauma
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Choi, Y.K.; Kim, K.W. Blood-neural barrier: Its diversity and coordinated cell-to-cell communication. BMB Rep. 2008, 41, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.D.; Egleton, R.D.; Davis, T.P. Molecular physiology and pathophysiology of tight junctions in the blood-brain barrier. Trends Neurosci. 2001, 24, 719–725. [Google Scholar] [CrossRef]
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.C.; Bath, M.F.; Stover, C.M.; Lambert, D.G.; Thompson, J.P. Exploring LPS-induced sepsis in rats and mice as a model to study potential protective effects of the nociceptin/orphanin FQ system. Peptides 2014, 61, 56–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K.A.; Kurihara, S.; Shibakusa, T.; Chiba, Y.; Mikami, T. Cystine improves survival rates in a LPS-induced sepsis mouse model. Clin. Nutr. 2015, 34, 1159–1165. [Google Scholar] [CrossRef] [PubMed]
- Ming, Z.; Sawicki, G.; Bekar, L.K. Acute systemic LPS-mediated inflammation induces lasting changes in mouse cortical neuromodulation and behavior. Neurosci. Lett. 2015, 590, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Erickson, M.A. The blood-brain barrier and immune function and dysfunction. Neurobiol. Dis. 2010, 37, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Salkeni, M.A.; Lynch, J.L.; Otamis-Price, T.; Banks, W.A. Lipopolysaccharide Impairs Blood-Brain Barrier P-glycoprotein Function in Mice Through Prostaglandin- and Nitric Oxide-Independent Pathways. J. Neuroimmune Pharmacol. 2009, 4, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Elwood, E.; Lim, Z.; Naveed, H.; Galea, I. The effect of systemic inflammation on human brain barrier function. Brain Behav. Immun. 2017, 62, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Qin, J.; Liu, M.; Ruan, Q.; Li, Y.; Zhang, Z. Role of Rho kinase in lysophosphatidic acid-induced altering of blood-brain barrier permeability. Int. J. Mol. Med. 2014, 33, 661–669. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Yin, F.; Omran, A.; Yang, L.; Xiang, Q.; Peng, J. PKC and RhoA signals cross-talk in Escherichia coli endotoxin induced alterations in brain endothelial permeability. Biochem. Biophys. Res. Commun. 2012, 425, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Gray, A.M.; Erickson, M.A.; Salameh, T.S.; Damodarasamy, M.; Sheibani, N.; Meabon, J.S.; Wing, E.E.; Morofuji, Y.; Cook, D.G.; et al. Lipopolysaccharide-induced blood-brain barrier disruption: Roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J. Neuroinflamm. 2015, 12, 223. [Google Scholar] [CrossRef] [PubMed]
- Niego, B.; Lee, N.; Larsson, P.; De Silva, T.M.; Au, A.E.; McCutcheon, F.; Medcalf, R.L. Selective inhibition of brain endothelial Rho-kinase-2 provides optimal protection of an in vitro blood-brain barrier from tissue-type plasminogen activator and plasmin. PLoS ONE 2017, 12, e0177332. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Jang, H.J.; Chung, T.W.; Jeong, S.I.; Cha, J.; Choi, J.Y.; Han, C.W.; Jang, Y.S.; Joo, M.; Jeong, H.S.; et al. Catalpol suppresses advanced glycation end-products-induced inflammatory responses through inhibition of reactive oxygen species in human monocytic THP-1 cells. Fitoterapia 2013, 86, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Jiang, B.; Zorn, A.; Zhao, R.G.; Liu, P.; An, L.J. Catalpol inhibits LPS plus IFN-gamma-induced inflammatory response in astrocytes primary cultures. Toxicol. In Vitro 2013, 27, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jin, C.; Li, Y.; Guan, S.; Han, F.; Zhang, S. Catalpol improves cholinergic function and reduces inflammatory cytokines in the senescent mice induced by d-galactose. Food Chem. Toxicol. 2013, 58, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, Q.; Dong, F.; Li, L.; Du, J.; Xie, Q.; Hu, H.; Yan, S.; Zhou, X.; Li, C.; et al. Catalpol downregulates vascular endothelialcadherin expression and induces vascular hyperpermeability. Mol. Med. Rep. 2016, 13, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, Z.; Jiang, Z.; Sun, L.; Ji, J.; Miao, J.; Zhang, X.; Li, X.; Huang, S.; Wang, T.; et al. Hypoglycemic effect of catalpol on high-fat diet/streptozotocin-induced diabetic mice by increasing skeletal muscle mitochondrial biogenesis. Acta Biochim. Biophys. Sin. 2014, 46, 738–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.F.; Li, D.Q.; Xue, H.Y.; Hu, B. Oral supplementation of catalpol ameliorates diabetic encephalopathy in rats. Brain Res. 2010, 1307, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.Y.; An, L.J.; Jiang, L.; Duan, Y.L.; Chen, J.; Jiang, B. Catalpol protects dopaminergic neurons from LPS-induced neurotoxicity in mesencephalic neuron-glia cultures. Life Sci. 2006, 80, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Li, X.; Jia, L.Q.; Wang, J.; Zhang, L.; Hou, D.; Wang, J.; Ren, L. Neuroprotective activities of catalpol against CaMKII-dependent apoptosis induced by LPS in PC12 cells. Br. J. Pharmacol. 2013, 169, 1140–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, A.; Hao, S.; Bi, J.; Bao, Y.; Zhang, X.; An, L.; Jiang, B. Effects of catalpol on mitochondrial function and working memory in mice after lipopolysaccharide-induced acute systemic inflammation. Exp. Toxicol. Pathol. 2009, 61, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Fu, K.; Piao, T.; Wang, M.; Zhang, J.; Jiang, J.; Wang, X.; Liu, H. Protective effect of catalpol on lipopolysaccharide-induced acute lung injury in mice. Int. Immunopharmacol. 2014, 23, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jia, R.; Wang, F.; Qiu, G.; Qiao, P.; Xu, X.; Wu, D. Catalpol protects mice against Lipopolysaccharide/D-galactosamine-induced acute liver injury through inhibiting inflammatory and oxidative response. Oncotarget 2017, 9, 3887–3894. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.P.; Pan, C.S.; Yan, L.; Liu, Y.Y.; Hu, B.H.; Chang, X.; Li, Q.; Huang, D.D.; Sun, H.Y.; Fu, G.; et al. Catalpol restores LPS-elicited rat microcirculation disorder by regulation of a network of signaling involving inhibition of TLR-4 and SRC. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G1091–G1104. [Google Scholar] [CrossRef] [PubMed]
- Wan, D.; Yang, X.; Wang, Y.; Zhu, H.; Wang, T.; Liu, Z.; Feng, S.; Fu, Y.; Wang, H.; Wang, J.; et al. Catalpol stimulates VEGF production via the JAK2/STAT3 pathway to improve angiogenesis in rats’ stroke model. J. Ethnopharmacol. 2016, 191, 169–179. [Google Scholar] [CrossRef]
- Xue, Q.; Liu, Y.; Qi, H.; Ma, Q.; Xu, L.; Chen, W.; Chen, G.; Xu, X. A Novel Brain Neurovascular Unit Model with Neurons, Astrocytes and Microvascular Endothelial Cells of Rat. Int. J. Biol. Sci. 2013, 9, 174–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniels, B.P.; Cruz-Orengo, L.; Pasieka, T.J.; Couraud, P.O.; Romero, I.A.; Weksler, B.; Cooper, J.A.; Doering, T.L.; Klein, R.S. Immortalized human cerebral microvascular endothelial cells maintain the properties of primary cells in an in vitro model of immune migration across the blood brain barrier. J. Neurosci. Methods 2013, 212, 173–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terry, S.; Nie, M.; Matter, K.; Balda, M.S. Rho signaling and tight junction functions. Physiology 2010, 25, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Amin, E.; Dubey, B.N.; Zhang, S.C.; Gremer, L.; Dvorsky, R.; Moll, J.M.; Taha, M.S.; Nagel-Steger, L.; Piekorz, R.P.; Somlyo, A.V.; et al. Rho-kinase: Regulation, (dys)function, and inhibition. Biol. Chem. 2013, 394, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M. Molecular anatomy of the brain endothelial barrier: An overview of the distributional features. Curr. Med. Chem. 2007, 14, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Essler, M.; Staddon, J.M.; Weber, P.C.; Aepfelbacher, M. Cyclic AMP blocks bacterial lipopolysaccharide-induced myosin light chain phosphorylation in endothelial cells through inhibition of Rho/Rho kinase signaling. J. Immunol. 2000, 164, 6543–6549. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, X.; Han, Y.; Li, C.; Chen, P.; Su, S.; Zhang, Y.; Pan, Z. Rho kinase inhibition by fasudil suppresses lipopolysaccharide-induced apoptosis of rat pulmonary microvascular endothelial cells via JNK and p38 MAPK pathway. Biomed. Pharmacother. 2014, 68, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.D.; Monsul, N.T.; Vender, J.R.; Lehmann, J.C. NMDA- and endothelin-1-induced increases in blood-brain barrier permeability quantitated with Lucifer yellow. J. Neurol. Sci. 1996, 136, 37–40. [Google Scholar] [CrossRef]
- Narushima, I.; Kita, T.; Kubo, K.; Yonetani, Y.; Momochi, C.; Yoshikawa, I.; Ohno, N.; Nakashima, T. Highly enhanced permeability of blood-brain barrier induced by repeated administration of endothelin-1 in dogs and rats. Pharmacol. Toxicol. 2003, 92, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Freeman, B.D.; Machado, F.S.; Tanowitz, H.B.; Desruisseaux, M.S. Endothelin-1 and its role in the pathogenesis of infectious diseases. Life Sci. 2014, 118, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenzie, J.A.; Ridley, A.J. Roles of Rho/ROCK and MLCK in TNF-alpha-induced changes in endothelial morphology and permeability. J. Cell. Physiol. 2007, 213, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Capaldo, C.T.; Nusrat, A. Cytokine regulation of tight junctions. Biochim. Biophys. Acta 2009, 1788, 864–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, Y.; Teng, T.; Li, R.; Simonyi, A.; Sun, G.Y.; Lee, J.C. TNFα alters occludin and cerebral endothelial permeability: Role of p38MAPK. PLoS ONE 2017, 12, e0170346. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Sun, Y.; Hu, J. Catalpol inhibits apoptosis in hydrogen peroxide-induced endothelium by activating the PI3K/Akt signaling pathway and modulating expression of Bcl-2 and Bax. Eur. J. Pharmacol. 2010, 628, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, Q.; Zhang, R.; Liu, S.; Xia, Z.; Hu, Y. Catalpol ameliorates beta amyloid-induced degeneration of cholinergic neurons by elevating brain-derived neurotrophic factors. Neuroscience 2009, 163, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xing, M.; Chen, W.; Zhang, J.; Qi, H.; Xu, X. HPLC-APCI-MS/MS method for the determination of catalpol in rat plasma and cerebrospinal fluid: Application to an in vivo pharmacokinetic study. J. Pharm. Biomed. Anal. 2012, 70, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Nemoto, K.; Ninomiya, N.; Kuno, M.; Kubota, M.; Yokota, H. Fasudil, a Rho-kinase inhibitor, attenuates lipopolysaccharide-induced vascular hyperpermeability and colonic muscle relaxation in guinea pigs. J. Surg. Res. 2012, 178, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Thorlacius, K.; Slotta, J.E.; Laschke, M.W.; Wang, Y.; Menger, M.D.; Jeppsson, B.; Thorlacius, H. Protective effect of fasudil, a Rho-kinase inhibitor, on chemokine expression, leukocyte recruitment, and hepatocellular apoptosis in septic liver injury. J. Leukoc. Biol. 2006, 79, 923–931. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, S.; Zou, L.; Wang, H.; He, R.; Liu, K.; Zhu, H. RhoA/ROCK-2 Pathway Inhibition and Tight Junction Protein Upregulation by Catalpol Suppresses Lipopolysaccaride-Induced Disruption of Blood-Brain Barrier Permeability. Molecules 2018, 23, 2371. https://doi.org/10.3390/molecules23092371
Feng S, Zou L, Wang H, He R, Liu K, Zhu H. RhoA/ROCK-2 Pathway Inhibition and Tight Junction Protein Upregulation by Catalpol Suppresses Lipopolysaccaride-Induced Disruption of Blood-Brain Barrier Permeability. Molecules. 2018; 23(9):2371. https://doi.org/10.3390/molecules23092371
Chicago/Turabian StyleFeng, Shan, Li Zou, Hongjin Wang, Ran He, Ke Liu, and Huifeng Zhu. 2018. "RhoA/ROCK-2 Pathway Inhibition and Tight Junction Protein Upregulation by Catalpol Suppresses Lipopolysaccaride-Induced Disruption of Blood-Brain Barrier Permeability" Molecules 23, no. 9: 2371. https://doi.org/10.3390/molecules23092371