3. Materials and Methods
3.1. General Experimental Methods
The reagents and solvents were used as obtained from the commercial sources. Compounds
3a [
14],
3b [
14], and
5b [
15], as well as benzyl azide (
8a) [
23], azidobenzene (
8b) [
11], and tetrazolo[1,5-
a]pyridine (
8c) [
24] were prepared as described in the literature. Column chromatography was carried out on Fluka Silica gel 60 (particle size 0.063–0.2 mm, activity acc. Brockmann and Schodder 2–3). Melting points were determined on the microscope hot stage, Kofler, PolyTherm, manufacturer Helmut Hund GmbH, Wetzlar and are uncorrected. TLC was carried out on pre-coated TLC sheets ALUGRAM
® SIL G/UV
254 for TLC, MACHEREY-NAGEL. NMR spectra were recorded with a Bruker Avance III 500 MHz NMR instrument operating at 500 MHz (
1H), 126 MHz (
13C) and 51 MHz (
15N) at 300 K. Proton spectra were referenced to TMS as internal standard, in some cases to the residual signal of DMSO-
d5 (at
δ 2.50 ppm) or CHCl
3 (at
δ 7.26 ppm). Carbon chemical shifts were determined relative to the
13C signal of DMSO-
d6 (39.52 ppm) or CDCl
3 (77.16 ppm).
15N chemical shifts were extracted from
1H–
15N
gs-HMBC spectra (with 20 Hz digital resolution in the indirect dimension and the parameters adjusted for a long-range
1H–
15N coupling constant of 5 Hz) determined with respect to external nitromethane and are corrected to external ammonia by addition of 380.5 ppm. Nitrogen chemical shifts are reported to one decimal place as measured of the spectrum, however, the data should not be considered to be more accurate than ±0.5 ppm because of the digital resolution limits of the experiment. Chemical shifts are given on the
δ scale (ppm). Coupling constants (
J) are given in Hz. Multiplicities are indicated as follows: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet) or br (broadened). Infrared spectra were recorded on FT-IR spectrometer Alpha (Bruker Optik GmbH Ettlingen, Ettlingen, Germany) using samples in potassium bromide disks and only the strongest/structurally most important peaks are listed. Electron impact mass spectra (EI) were recorded on a Shimadzu QP–2010 instrument at 70 eV. HRMS spectra were recorded with Agilent 6224 Accurate Mass TOF LC/MS system with electrospray ionization (ESI). Elemental analyses (C, H, N) were performed with FlashEA1112 Automatic Elemental Analyser (Thermo Fisher Scientific Inc., Waltham, MA, USA).
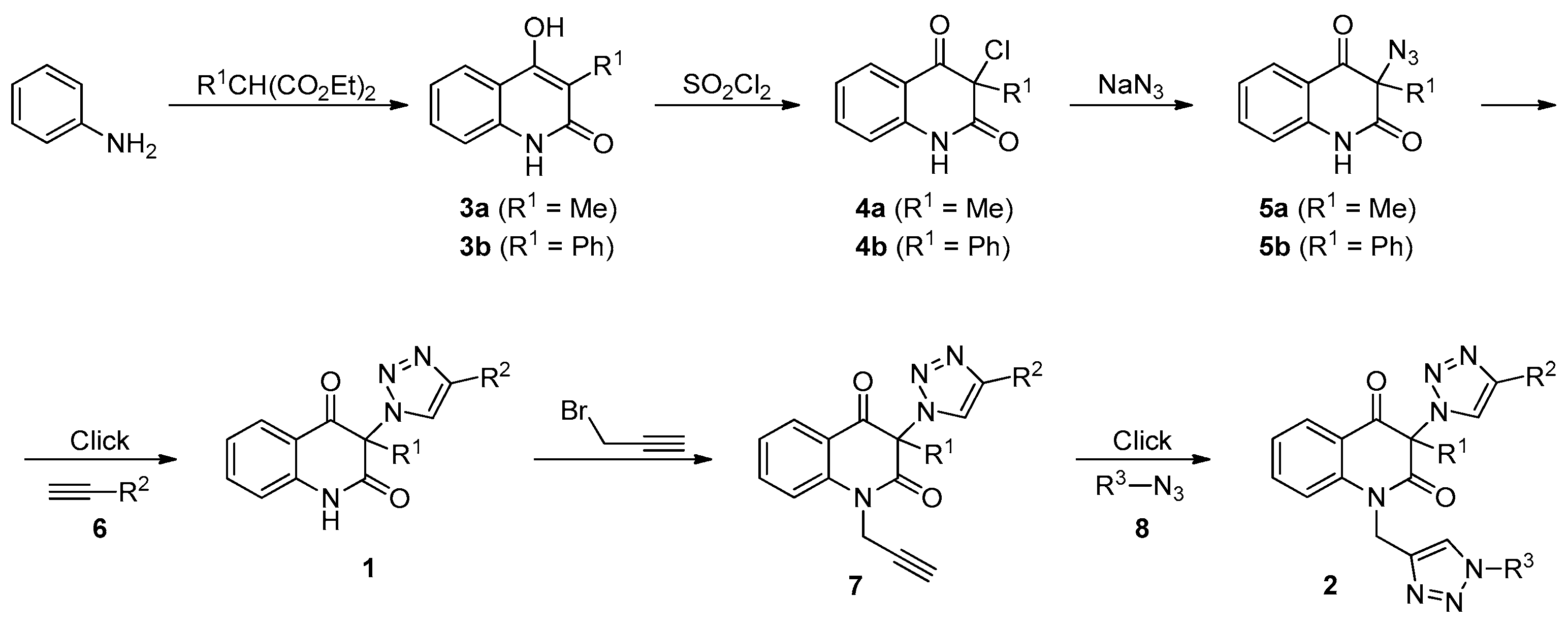
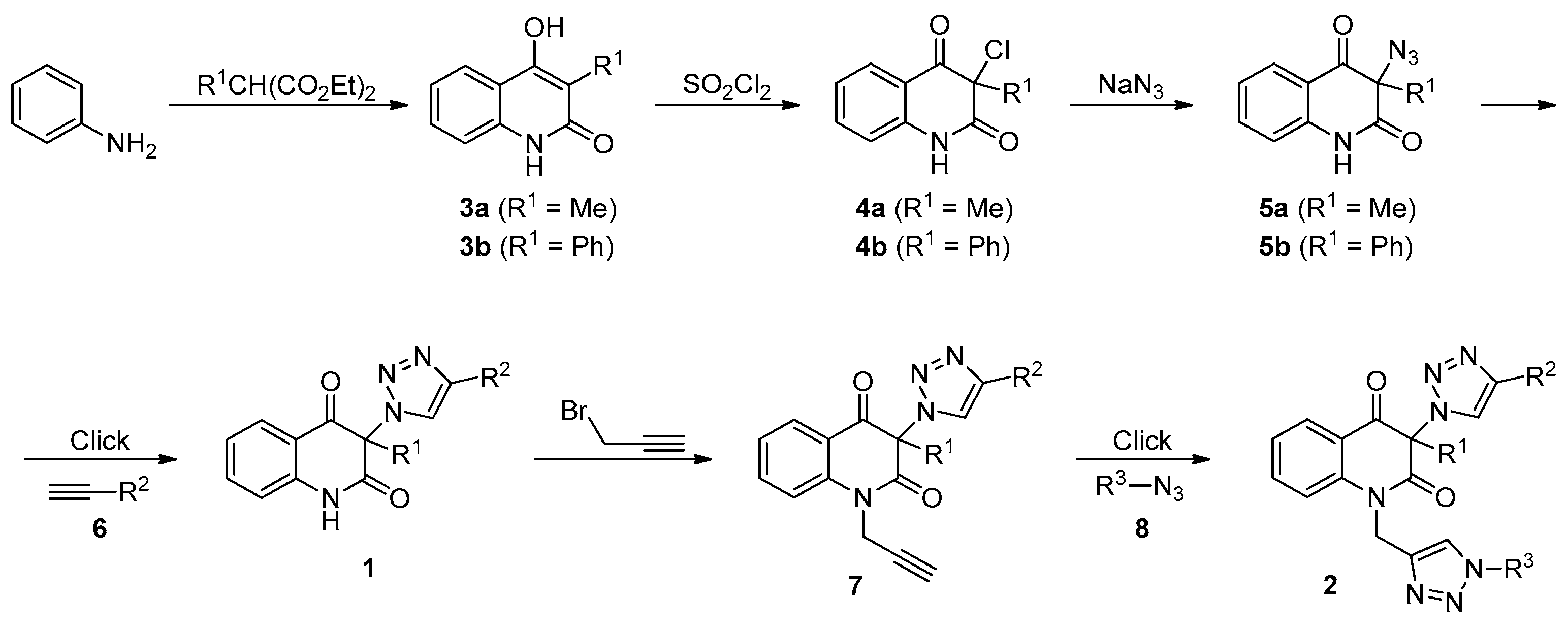
3.2. General Procedure for the Synthesis of 3-Chloroquinoline-2,4(1H,3H)-Diones 4 (Scheme 1)
The 3-Chloroquinoline-2,4(1
H,3
H)-diones
4a [
15] and
4b [
16], were prepared from 4‑hydroxyquinolin-2(1
H)-ones
3a [
14] and
3b [
14], respectively, according to the procedures described in the literature.
3-Chloro-3-methylquinoline-2,4(1H,3H)-dione (4a). Compound
4a (19.71 g, 94.0 mmol, 94%) was prepared from
3a (17.52 g, 100 mmol). Yellow crystals, m.p. 178–181 °C (benzene), m.p. [
15] 172 °C (acetic acid—water);
Rf = 0.52 (30% ethyl acetate in chloroform);
1H NMR (500 MHz, CDCl
3)
δ 1.99 (s, 3H), 7.06 (d, 1H,
J = 8.0 Hz), 7.22 (dd, 1H,
J = 7.6, 7.6 Hz), 7.59–7.66 (m, 1H), 8.02 (d, 1H,
J = 7.7 Hz), 9.41 (s, 1H);
13C NMR (126 MHz, CDCl
3)
δ 21.2, 62.8, 116.7, 118.1, 124.5, 129.1, 136.8, 139.6, 169.2, 188.4; IR (cm
−1):
ν 3203, 3072, 3004, 2940, 1709, 1674, 1614, 1600, 1486, 1439, 1379, 1239, 770, 440; MS (EI)
m/
z (%): 212 (4, [M + 3]
+), 211 (33, [M (
37Cl)]
+), 210 (17, [M + 1]
+), 209 (100, [M (
35Cl)]
+), 208 (18), 175 (15), 174 (36), 146 (68), 128 (17), 120 (18), 119 (59), 92 (32), 91 (15); HRMS (ESI+):
m/
z calcd for C
10H
9ClNO
2+ [M + H]
+ 210.0316, found 210.0313. Anal. Calcd for C
10H
8ClNO
2 (209.63): C, 57.30; H, 3.85; N, 6.68%. Found: C, 57.18; H, 3.83; N, 6.61%.
3-Chloro-3-phenylquinoline-2,4(1H,3H)-dione (4b). Compound
4b (26.08 g, 96.0 mmol, 96%) was prepared from
3b (23.73 g, 100 mmol). Pale yellow needles, m.p. 182–185 °C (benzene), m.p. [
16] 178–180 °C (ethanol);
Rf = 0.57 (30% ethyl acetate in chloroform).
1H NMR (500 MHz, CDCl
3)
δ 7.04 (d, 1H,
J = 8.0 Hz, H-8), 7.18 (ddd, 1H,
J = 7.8, 7.4, 0.7 Hz, H-6), 7.33–7.39 (m, 3H, H‑3
C, H‑4
C, H‑5
C), 7.51–7.54 (m, 2H, H-2
C, H-6
C), 7.55 (ddd, 1H,
J = 7.3, 6.5, 1.5 Hz, H-7), 7.97 (dd, 1H,
J = 7.8, 1.2 Hz, H-5), 9.82 (s, 1H, H-1);
13C NMR (126 MHz, CDCl
3)
δ 74.9 (C-3), 116.9 (C-8), 118.7 (C-4a), 124.7 (C-6), 127.4 (C-2
C, C-6
C), 129.1 (C-5), 129.2 (C-3
C, C-5
C), 129.8 (C-4
C), 134.6 (C-1
C), 137.0 (C-7), 139.4 (C-8a), 168.8 (C-2), 187.9 (C-4); IR (cm
−1):
ν 3201, 3138, 3082, 2992, 2926, 1716, 1680, 1613, 1595, 1485, 1365, 755, 743, 690; MS (EI)
m/
z (%): 273 (7, [M (
37Cl)]
+), 271 (21, [M (
35Cl)]
+), 238 (12), 237 (80), 236 (100), 218 (10), 120 (63), 119 (19), 92 (34), 89 (10), 77 (12), 76 (10), 65 (14), 63 (10); HRMS (ESI+):
m/
z calcd for C
15H
11ClNO
2+ [M + H]
+ 272.0473, found 272.0480. Anal. Calcd for C
15H
10ClNO
2 (271.70): C, 66.31; H, 3.71; N, 5.16%. Found: C, 66.07; H, 3.62; N, 5.29%.
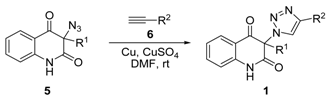
3.3. General Procedure for the Synthesis of 3-Azidoquinoline-2,4(1H,3H)-Diones 5 (Scheme 1)
To a stirred solution of the 3-chloroquinoline-2,4(1H,3H)-dione 4 (40 mmol) in DMF (200 mL), sodium azide (3.90 g, 60 mmol) was added in small portions during 10 min. The reaction mixture was stirred in darkness for additional 2 h and then poured into ice-water (1.5 L). The precipitated solid was filtered, washed with water and dried at 50 °C in darkness, which afforded product 5, pure according to TLC and 1H NMR spectrum, which was crystallized from benzene.
3-Azido-3-methylquinoline-2,4(1H,3H)-dione (5a). Compound 5a (8.47 g, 39.2 mmol, 98%) was prepared from 4a (8.39 g, 40.0 mmol). Colorless needles, m.p. 158–161 °C (benzene, 87% yield of recrystallization); Rf = 0.30 (30% ethyl acetate in chloroform). 1H NMR (500 MHz, CDCl3) δ 1.86 (s, 3H, CH3), 7.11 (d, 1H, J = 8.0 Hz, H-8), 7.22 (dd, 1H, J = 7.4, 7.4 Hz, H-6), 7.60–7.67 (m, 1H, H-7), 7.98 (d, 1H, J = 7.3 Hz, H-5), 9.86 (s, 1H, H-1); 13C NMR (126 MHz, CDCl3) δ 23.6 (CH3), 70.0 (C-3), 116.9 (C-8), 118.0 (C-4a), 124.6 (C-6), 128.6 (C-5), 137.2 (C-7), 140.0 (C-8a), 171.6 (C-2), 191.7 (C-4); IR (cm−1): ν 3202, 3078, 3005, 2936, 2108, 1708, 1682, 1614, 1598, 1485, 1392, 1284, 1156, 755, 612; MS (EI) m/z (%): 217 (0.24, [M + 1]+), 216 (2, [M]+), 147 (15), 120 (11), 119 (100), 92 (35), 91 (11), 64 (12); HRMS (ESI+): m/z calcd for C10H9N4O2+ [M + H]+ 217.0720, found 217.0724. Anal. Calcd for C10H8N4O2 (216.20): C, 55.55; H, 3.73; N, 25.91%. Found: C, 55.44; H, 3.72; N, 25.98%.
3-Azido-3-phenylquinoline-2,4(1H,3H)-dione (5b). Compound
5b (10.90 g, 39.2 mmol, 98%) was prepared from
4b (10.87 g, 40.0 mmol). Colorless needles, m.p. 186–189 °C (benzene, 96% yield of recrystallization); m.p. [
9] 173–181 °C (benzene);
Rf = 0.33 (38% ethyl acetate in petroleum ether);
1H NMR (500 MHz, CDCl
3)
δ 6.98 (d, 1H,
J = 8.1 Hz, H-8), 7.16 (dd, 1H,
J = 7.6, 7.6 Hz, H-6), 7.38–7.43 (m, 3H, H-3
C, H-4
C, H-5
C), 7.48–7.53 (m, 2H, H-2
C, H-6
C), 7.54 (ddd, 1H,
J = 7.7, 7.7, 1.6 Hz, H-7), 7.93 (dd, 1H,
J = 7.8, 1.6 Hz, H-5), 9.30 (s, 1H, H-1);
13C NMR (126 MHz, CDCl
3)
δ 78.0 (C-3), 116.7 (C-8), 119.5 (C-4a), 124.6 (C-6), 127.3 (C-2
C, C-6
C), 128.6 (C-5), 129.8 (C-3
C, C-5
C), 130.4 (C-4
C), 132.6 (C-1
C), 136.9 (C-7), 139.4 (C-8a), 170.2 (C-2), 189.9 (C-4);
15N NMR (51 MHz, CDCl
3)
δ 133.4 (N-1); IR (cm
−1):
ν 3244, 2105, 1718, 1705, 1685, 1611, 1484, 1356, 1256, 877, 773, 744, 702, 611, 525; MS (EI)
m/
z (%): 250 (7, [M − N
2]
+), 236 (8, [M − N
3]
+), 147 (28), 120 (14), 119 (100), 104 (15), 92 (32), 77 (10), 76 (10), 64 (14); HRMS (ESI+):
m/
z calcd for C
15H
11N
2O
2+ [M − N
2 + H]
+ 251.0815, found 251.0818. HRMS (ESI−):
m/
z calcd for C
15H
9N
4O
2− [M − H]
− 277.0731, found 277.0732; calcd for C
15H
9N
2O
2− [M − N
2 − H]
− 249.0670, found 249.0671. Anal. Calcd for C
15H
10N
4O
2 (278.27): C, 64.74; H, 3.62; N, 20.13%. Found: C, 64.54; H, 3.56; N, 20.38%.
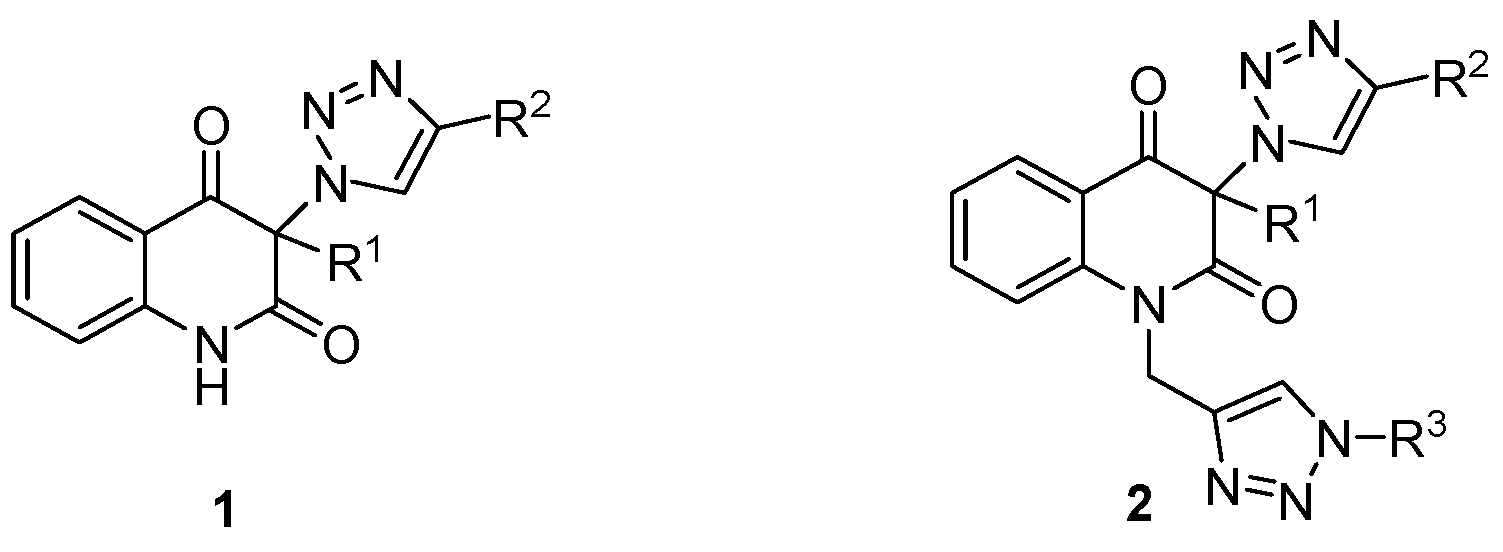
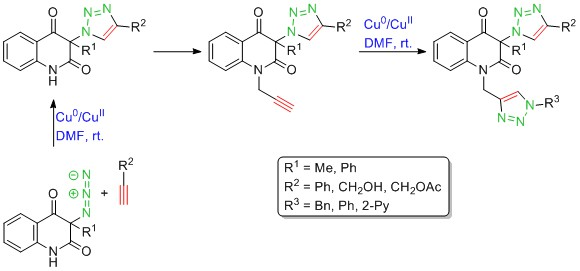
3.4. General Procedure for the Synthesis of 3-(1H-1,2,3-Triazol-1-yl)Quinoline-2,4(1H,3H)-Diones 1a–d by Employing CuSO4/Cu0/DMF Conditions (Table 2, Entries 1 and 3–5)
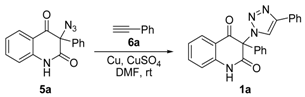
A solution of phenylacetylene (6a) (1.287 g, 12.6 mmol) or propargyl alcohol (6b) (706 mg, 12.6 mmol) in DMF (4 mL) was added dropwise to a vigorously stirred mixture of 3-azidoquinoline-2,4(1H,3H)-dione 5 (12 mmol), CuSO4⋅5H2O (300 mg, 1.2 mmol), granular copper (1.5 g, 24 mmol) and DMF (24 mL). The reaction mixture was stirred in darkness for 30 min. Afterward, (NH4)2CO3 (3.5 g, 36 mmol) and water (12 mL) were added to the resulting brown-black suspension and the stirring was continued for 10 min. The reaction mixture was subjected to column chromatography with silica gel (15 g, column diameter of 1 cm) as a stationary phase, and 10% ethanol in chloroform as a mobile phase. Combined fractions containing yellow eluate were washed with saturated aqueous NH4Cl (5 × 50 mL), dried (Na2SO4), and concentrated under reduced pressure to afford pure products 1a–d, which were recrystallized from ethanol for analyses.
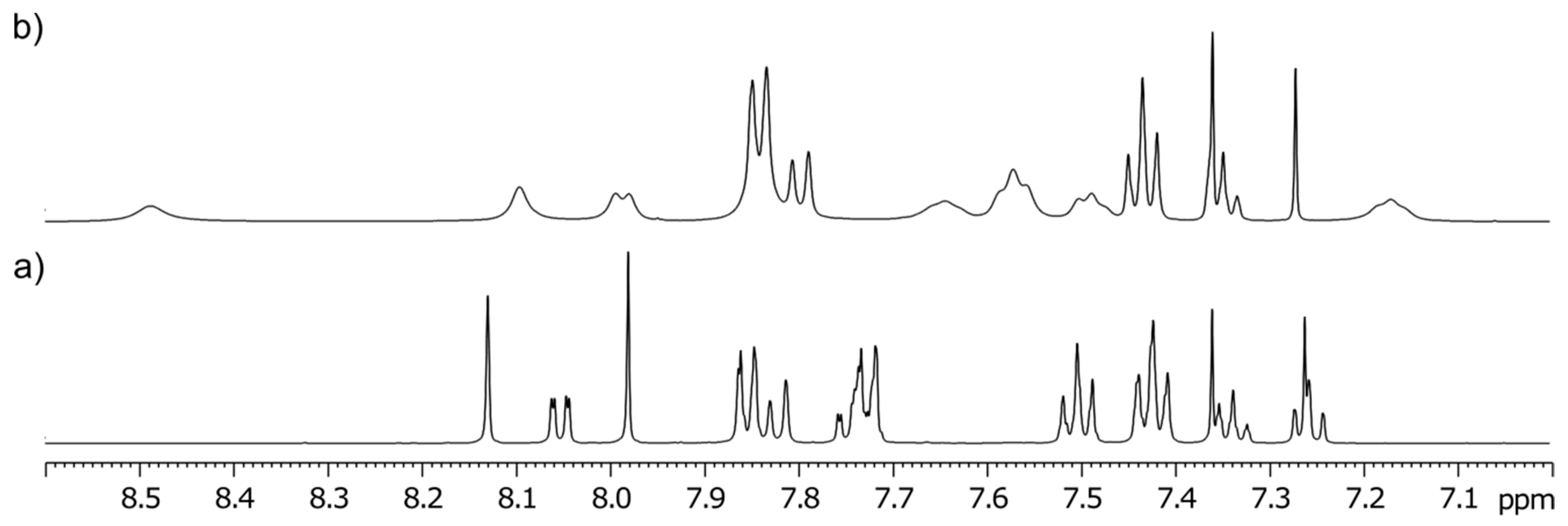
3-Methyl-3-(4-phenyl-1H-1,2,3-triazol-1-yl)quinoline-2,4(1H,3H)-dione (1a). Colorless solid, m.p. 217–219 °C (ethanol); Rf = 0.35 (30% ethyl acetate in chloroform); 1H NMR (500 MHz, DMSO-d6) δ 2.15 (s, 3H, CH3), 7.22–7.27 (m, 2H, H-6, H-8), 7.33–7.39 (m, 1H, H-4B), 7.45–7.50 (m, 2H, H-3B, H-5B), 7.73–7.79 (m, 1H, H-7), 7.83–7.90 (m, 3H, H-5, H-2B, H-6B), 8.89 (s, 1H, H-5A), 11.48 (s, 1H, H-1); 13C NMR (126 MHz, DMSO-d6) δ 23.1 (CH3), 72.2 (C-3), 117.0 (C-8), 117.4 (C-4a), 122.4 (C-5A), 123.5 (C-6), 125.1 (C-2B, C-6B), 127.7 (C-5), 128.0 (C-4B), 129.0 (C-3B, C-5B), 130.6 (C-1B), 137.3 (C-7), 141.6 (C-8a), 145.8 (C-4A), 168.5 (C-2), 190.7 (C-4); IR (cm−1): ν 3137, 2911, 1714, 1683, 1612, 1483, 1430, 1386, 1355, 1238, 1023, 808, 759, 690, 594; MS (EI) m/z (%): 319 (2, [M + 1]+), 318 (8, [M]+), 117 (14), 116 (100), 102 (12), 89 (14); HRMS (ESI+): m/z calcd for C18H15N4O2+ [M + H]+ 319.1190, found 319.1188. Anal. Calcd for C18H14N4O2 (318.33): C, 67.91; H, 4.43; N, 17.60%. Found: C, 67.80; H, 4.47; N, 17.89%.
3-Phenyl-3-(4-phenyl-1H-1,2,3-triazol-1-yl)quinoline-2,4(1H,3H)-dione (1b). Colorless solid, m.p. 280–283 °C (ethanol); m.p. [
9] 274–277 °C (ethanol);
Rf = 0.37 (30% ethyl acetate in chloroform);
1H NMR (500 MHz, DMSO-
d6)
δ 7.12 (d, 1H,
J = 8.1 Hz, H-8), 7.16–7.20 (m, 1H, H-6), 7.31–7.37 (m, 1H, H-4
B), 7.40–7.47 (m, 4H, H-3
B, H-5
B, H-2
C, H-6
C), 7.49–7.55 (m, 3H, H-3
C, H-4
C, H-5
C), 7.62–7.67 (m, 1H, H-7), 7.80–7.84 (m, 2H, H-2
B, H-6
B), 7.86 (dd, 1H,
J = 7.8, 1.4 Hz, H-5), 8.49 (s, 1H, H-5
A), 11.68 (s, 1H, H-1);
13C NMR (126 MHz, DMSO-
d6)
δ 80.0 (C-3), 116.7 (C-8), 119.2 (C-4a), 123.4 (C-5
A), 123.5 (C-6), 125.2 (C-2
B, C-6
B), 127.6 (C-5), 128.0 (C-4
B), 128.9 (C-2
C, C-6
C), 129.0 (C-3
B, C-5
B), 129.6 (C-3
C, C-5
C), 129.9 (C-1
C), 130.5 (C-1
B), 130.6 (C-4
C), 137.0 (C-7), 140.5 (C-8a), 145.3 (C-4
A), 166.8 (C-2), 188.9 (C-4); IR (cm
−1):
ν 3275, 3169, 1721, 1690, 1613, 1595, 1486, 1452, 1353, 854, 771, 756, 699, 666, 607; MS (EI)
m/
z (%): 381 (2, [M + 1]
+), 380 (8, [M]
+), 247 (13), 237 (15), 236 (56), 220 (13), 218 (13), 120 (11), 117 (10), 116 (100), 102 (15), 92 (10), 89 (15), 77 (14); HRMS (ESI+):
m/
z calcd for C
23H
17N
4O
2+ [M + H]
+ 381.1346, found 381.1341. Anal. Calcd for C
23H
16N
4O
2 (380.40): C, 72.62; H, 4.24; N, 14.73%. Found: C, 72.40; H, 4.23; N, 14.90%.
3-(4-(Hydroxymethyl)-1H-1,2,3-triazol-1-yl)-3-methylquinoline-2,4(1H,3H)-dione (1c). Colorless solid, m.p. 188–189 °C (ethanol); Rf = 0.35 (30% ethyl acetate in chloroform); 1H NMR (500 MHz, DMSO-d6) δ 2.08 (s, 3H, CH3), 4.55 (d, 2H, J = 5.6 Hz, CH2), 5.28 (t, 1H, J = 5.6 Hz, OH), 7.18–7.25 (m, 2H, H-6, H-8), 7.69–7.76 (m, 1H, H-7), 7.83 (dd, 1H, J = 8.1, 1.4 Hz, H-5), 8.26 (s, 1H, H-5A), 11.39 (s, 1H, H-1); 13C NMR (126 MHz, DMSO-d6) δ 23.1 (CH3), 55.0 (CH2), 71.9 (C-3), 116.9 (C-8), 117.5 (C-4a), 123.3 (C-6), 123.7 (C-5A), 127.6 (C-5), 137.1 (C-7), 141.6 (C-8a), 147.4 (C-4A), 168.7 (C-2), 190.8 (C-4); IR (cm−1): ν 3148, 2992, 2919, 1729, 1682, 1613, 1486, 1378, 1345, 1235, 1189, 1009, 751, 667, 590; MS (EI) m/z (%): 273 (2, [M + 1]+), 272 (13, [M]+), 185 (68), 175 (89), 174 (45), 146 (100), 128 (58), 120 (70), 119 (75), 92 (66), 91 (39), 77 (39), 65 (37), 55 (39), 42 (79); HRMS (ESI+): m/z calcd for C13H13N4O3+ [M + H]+ 273.0982, found 273.0981. Anal. Calcd for C13H12N4O3 (272.26): C, 57.35; H, 4.44; N, 20.58%. Found: C, 57.20; H, 4.42; N, 20.83%.
3-(4-(Hydroxymethyl)-1H-1,2,3-triazol-1-yl)-3-phenylquinoline-2,4(1H,3H)-dione dimethylformamide solvate (1d·DMF). Colorless solid, m.p. 139–143 °C (ethanol); m.p. [
9] 116–135 °C (benzene);
Rf = 0.27 (10% ethanol in chloroform);
1H NMR (500 MHz, DMSO-
d6)
δ 4.53 (d, 2H,
J = 5.7 Hz, CH
2), 5.22 (t, 1H,
J = 5.7 Hz, OH), 7.09 (d, 1H,
J = 8.1 Hz, H-8), 7.13–7.18 (m, 1H, H-6), 7.36–7.42 (m, 2H, H-2
C, H-6
C), 7.47–7.53 (m, 3H, H-3
C, H-4
C, H-5
C), 7.59–7.65 (m, 1H, H-7), 7.77 (s, 1H, H-5
A), 7.83 (dd, 1H,
J = 7.8, 1.3 Hz, H-5), 11.60 (s, 1H, H-1);
13C NMR (126 MHz, DMSO-
d6)
δ 55.0 (CH
2), 79.7 (C-3), 116.7 (C-8), 119.2 (C-4a), 123.4 (C-6), 124.8 (C-5
A), 127.5 (C-5), 128.8 (C-2
C, C-6
C), 129.6 (C-3
C, C-5
C), 130.2 (C-1
C), 130.5 (C-4
C), 136.9 (C-7), 140.6 (C-8a), 146.8 (C-4
A), 166.8 (C-2), 189.0 (C-4); IR (cm
−1):
ν 3392, 3136, 2926, 1724, 1692, 1654, 1613, 1485, 1438, 1353, 857, 769, 752, 665, 603; MS (EI)
m/
z (%): 335 (0.8, [M + 1]
+), 334 (4, [M]
+), 305 (37), 275 (18), 249 (30), 247 (27), 237 (50), 236 (100), 218 (35), 208 (18), 180 (20), 120 (33), 104 (23), 92 (23), 77 (34); HRMS (ESI+):
m/
z calcd for C
18H
15N
4O
3+ [M + H]
+ 335.1139, found 335.1138. Anal. Calcd for C
21H
21N
5O
4 (407.42): C, 61.91; H, 5.20; N, 17.19%. Found: C, 61.89; H, 5.24; N, 17.28%.
3.5. Synthesis of Compound 1a by Employing CuSO4∙5H2O/l-Ascorbic Acid/CH2Cl2/Water Conditions (Table 2, Entry 2)
To a solution of azide 5a (162 mg, 0.75 mmol) and phenylacetylene (6a) (153 mg, 1.5 mmol) in dichloromethane (8 mL) a solution of l-ascorbic acid (106 mg, 0.60 mmol) in water (4 mL), and a solution of CuSO4∙5H2O (15 mg, 0.06 mmol) in water (4mL) were added. The two-phase reaction mixture was stirred in darkness at room temperature for 48 h. The reaction mixture was extracted with chloroform (5 × 30 mL). The combined organic layers were dried (Na2SO4), filtered, and evaporated to dryness. The residue was dissolved in chloroform (5 mL) and subjected to silica gel (30 g) column chromatography using 38% ethyl acetate in hexane as eluent, affording compound 1a (199 mg, 63 mmol 83%).
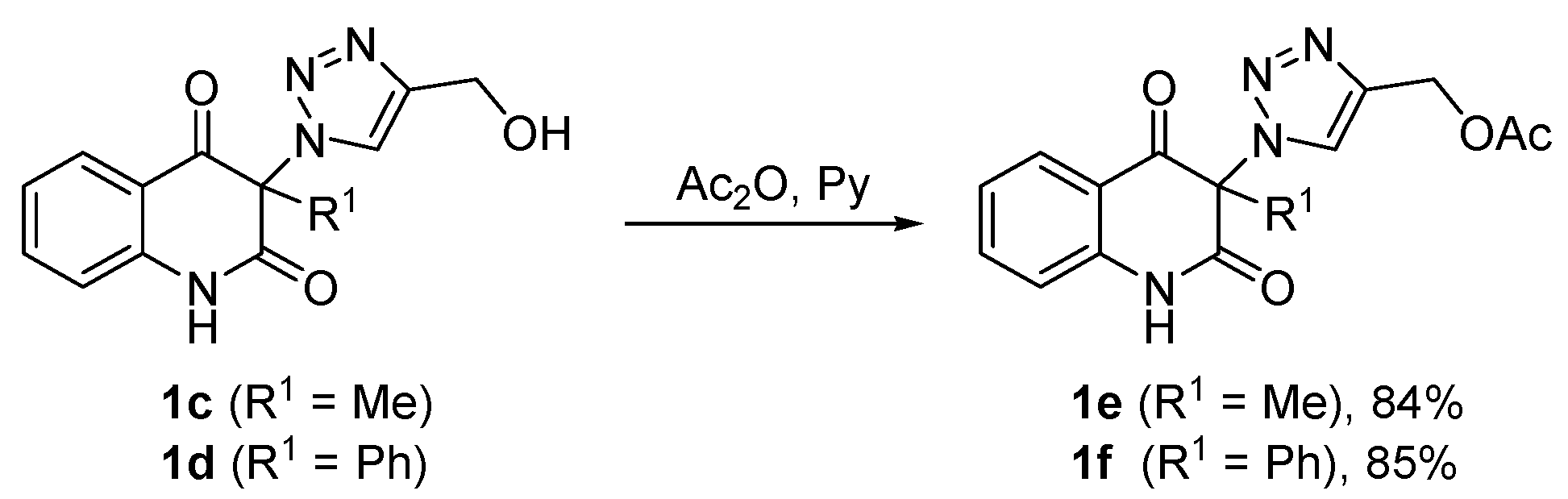
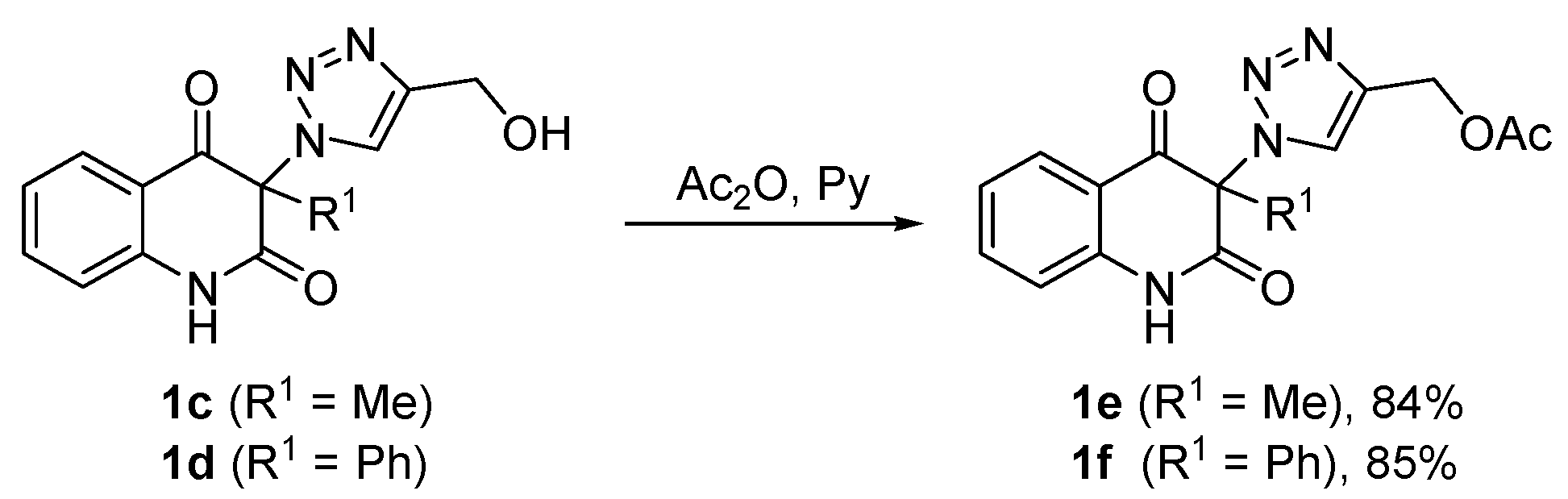
3.6. General Procedure for the Synthesis of (1-(2,4-Dioxo-1,2,3,4-Tetrahydroquinolin-3-yl)-1H-1,2,3-Triazol-4-yl)Methyl Acetates 1e,f (Scheme 2)
Acetic anhydride (12 mL, 12.9 g, 126 mmol) was added to a light yellow solution of compound 1c or 1d (6 mmol) in pyridine (18 mL) under stirring during 2 min. The reaction mixture was stirred for 1 h, followed by evaporation of volatiles under reduced pressure. The remaining pyridine was removed by co-distillation with ethanol (6 × 40 mL). The residue was triturated with water (300 mL) to form a white precipitate which was collected by filtration on a sintered glass filter with suction, washed with water to neutral and dried to give acetates 1e or 1f. The crude product was recrystallized from the solvent indicated below.
(1-(3-Methyl-2,4-dioxo-1,2,3,4-tetrahydroquinolin-3-yl)-1H-1,2,3-triazol-4-yl)methyl acetate (1e). Compound 1e (1.58 g, 5.04 mmol, 84%) was prepared from 1c (1.63 g, 6.0 mmol). Pale yellow solid, m.p. 145–148 °C (ethyl acetate); Rf = 0.33 (5% ethanol in chloroform); 1H NMR (500 MHz, DMSO-d6) δ 2.06 (s, 3H, COCH3), 2.09 (s, 3H, C-3–CH3), 5.16 (s, 2H, CH2), 7.19–7.26 (m, 2H, H-6, H-8), 7.70–7.77 (m, 1H, H-7), 7.83 (dd, 1H, J = 8.0, 1.4 Hz, H-5), 8.45 (s, 1H, H-5A), 11.40 (s, 1H, H-1); 13C NMR (126 MHz, DMSO-d6) δ 20.6 (COCH3), 23.2 (C-3–CH3), 57.1 (CH2), 72.4 (C-3), 116.9 (C-8), 117.5 (C-4a), 123.3 (C-6), 125.8 (C-5A), 127.6 (C-5), 137.1 (C-7), 141.4 (C-4A), 141.6 (C-8a), 168.6 (C-2), 170.1 (COCH3), 190.7 (C-4); 15N NMR (51 MHz, DMSO-d6) δ 133.5 (N-1), 248.7 (N-1A), 354.1 (N-3A), 362.9 (N-2A); IR (cm−1): ν 3467, 3249, 3148, 2920, 1722, 1685, 1613, 1485, 1439, 1384, 1355, 1239, 1028, 759, 666; MS (EI) m/z (%): 315 (2, [M + 1]+), 314 (11, [M]+), 244 (22), 201 (22), 175 (71), 174 (31), 146 (43), 128 (26), 120 (25), 119 (27), 92 (24), 55 (20), 43 (100), 42 (26); HRMS (ESI+): m/z calcd for C15H15N4O4+ [M + H]+ 315.1088, found 315.1087. Anal. Calcd for C15H14N4O4 (314.30): C, 57.32; H, 4.49; N, 17.83%. Found: C, 57.32; H, 4.59; N, 17.58%.
(1-(2,4-Dioxo-3-phenyl-1,2,3,4-tetrahydroquinolin-3-yl)-1H-1,2,3-triazol-4-yl)methyl acetate (1f). Compound 1f (1.92 g, 5.1 mmol, 85%) was prepared from 1d (2.01 g, 6.0 mmol). Colorless crystals, m.p. 130–134 °C (ethanol, 80% yield of recrystallization); Rf = 0.40 (5% ethanol in chloroform); 1H NMR (500 MHz, DMSO-d6) δ 2.04 (s, 3H, CH3), 5.13 (s, 2H, CH2), 7.09 (d, 1H, J = 8.1 Hz, H-8), 7.13–7.18 (m, 1H, H-6), 7.35–7.42 (m, 2H, H-2C, H-6C), 7.46–7.54 (m, 3H, H-3C, H-4C, H-5C), 7.59–7.65 (m, 1H, H-7), 7.83 (dd, 1H, J = 7.8, 1.3 Hz), 8.07 (s, 1H, H-5A), 11.62 (s, 1H, H-1); 13C NMR (126 MHz, DMSO-d6) δ 20.7 (CH3), 57.1 (CH2), 79.9 (C-3), 116.7 (C-8), 119.3 (C-4a), 123.4 (C-6), 127.0 (C-5A), 127.5 (C-5), 128.8 (C-2C, C-6C), 129.6 (C-3C, C-5C), 130.0 (C-1C), 130.6 (C-4C), 136.9 (C-7), 140.5 (C-8a), 140.8 (C-4A), 166.8 (C-2), 170.1 (COCH3), 188.8 (C-4); IR (cm–1): ν 3501, 3155, 2920, 1722, 1707, 1686, 1614, 1594, 1484, 1358, 1252, 1229, 1063, 857, 759; MS (EI) m/z (%): 377 (1, [M + 1]+), 376 (6, [M]+), 306 (16), 289 (18), 288 (54), 263 (15), 237 (50), 236 (100), 218 (34), 180 (14), 141 (14), 120 (24), 92 (14), 77 (19), 43 (16); HRMS (ESI+): m/z calcd for C20H17N4O4+ [M + H]+ 377.1244, found 377.1241.
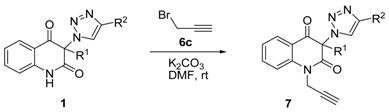
3.7. General Procedure for the Synthesis of 3-(1H-1,2,3-Triazol-1-yl)-1-(prop-2-yn-1-yl)Quinoline-2,4(1H,3H)-Diones 7 (Table 3)
The mixture of the appropriate compound
1a,
b,
e,
f (8.00 mmol), potassium carbonate (3.32 g, 24 mmol), and DMF (40 mL) was stirred for 40 min. During this time, the original yellow color of the suspension changed to orange. Afterwards, under continued stirring, an 80% solution of propargyl bromide (
6c) in toluene (1.78 g, 12 mmol) diluted with DMF (20 mL) was added dropwise during 1 min and stirring was continued for 90 min. Then the mixture was reduced
in vacuo and then toluene (50 mL) was added and the whole evaporated
in vacuo at 50 °C. This was repeated seven times in order to remove traces of DMF. The residual light brown solid was suspended in chloroform (100 mL) and the suspension was acidified with 0.5
m HCl, whereas carbon dioxide was evolved owing to decomposition of unreacted potassium carbonate. The formed emulsion was diluted with water, organic phase was separated and aqueous phase was extracted with chloroform (5 × 40 mL). The organic phases were combined, dried (Na
2SO
4), filtered and taken down
in vacuo. The residual solid TLC pure product was crystallized from a suitable solvent. The yields of prepared compounds
7 are given in
Table 3.
3-Methyl-3-(4-phenyl-1H-1,2,3-triazol-1-yl)-1-(prop-2-yn-1-yl)quinoline-2,4(1H,3H)-dione (7a). Colorless crystals, m.p. 187–189 °C (benzene); Rf = 0.63 (30% ethyl acetate in chloroform); 1H NMR (500 MHz, DMSO-d6) δ 2.16 (s, 3H, CH3), 3.39 (dd, 1H, J = 2.3, 2.3 Hz, C≡CH), 4.90 (dd, 1H, J = 18.1, 2.3 Hz, N-1–CHα), 4.97 (dd, 1H, J = 18.1, 2.3 Hz, N-1–CHβ), 7.34–7.42 (m, 2H, H-6, H-4B), 7.48 (dd, 2H, J = 7.7, 7.7 Hz, H-3B, H-5B), 7.61 (d, 1H, J = 8.4 Hz, H-8), 7.84–7.89 (m, 2H, H-2B, H-6B), 7.89–7.95 (m, 1H, H-7), 8.00 (dd, J = 7.7, 1.5 Hz, H-5), 8.89 (s, 1H, H-5A); 13C NMR (126 MHz, DMSO-d6) δ 23.3 (CH3), 32.7 (N-1–CH2), 72.6 (C-3), 75.4 (C≡CH), 78.2 (C≡CH), 116.7 (C-8), 119.0 (C-4a), 122.5 (C-5A), 124.2 (C-6), 125.1 (C-2B, C-6B), 128.1 (C-4B), 128.2 (C-5), 129.0 (C-3B, C-5B), 130.5 (C-1B), 137.3 (C-7), 140.8 (C-8a), 145.9 (C-4A), 167.7 (C-2), 189.7 (C-4); IR (cm−1): ν 3261, 3173, 2122, 1713, 1678, 1601, 1469, 1427, 1381, 1368, 1353, 1306, 1189, 769, 754; MS (EI) m/z (%): 357 (2, [M + 1]+), 356 (8, [M]+), 259 (10), 128 (11), 117 (16), 116 (100), 102 (17), 90 (11), 89 (16), 77 (10), 76 (10); HRMS (ESI+): m/z calcd for C21H17N4O2+ [M + H]+ 357.1346, found 357.1342. Anal. Calcd for C21H16N4O2 (356.38): C, 70.77; H, 4.53; N, 15.72%. Found: C, 70.81; H, 4.58; N, 15.82%.
3-Phenyl-3-(4-phenyl-1H-1,2,3-triazol-1-yl)-1-(prop-2-yn-1-yl)quinoline-2,4(1H,3H)-dione (7b). Colorless crystals, m.p. 232–234 °C (ethanol); Rf = 0.69 (30% ethyl acetate in chloroform); 1H NMR (500 MHz, CDCl3) δ 2.34 (dd, 1H, J = 2.4, 2.4 Hz), 4.51 (dd, 1H, J = 17.8, 2.3 Hz), 5.37 (dd, 1H, J = 17.8, 2.3 Hz), 7.23 (dd, 1H, J = 7.6, 7.6 Hz), 7.26 (s, 1H), 7.27–7.31 (m, 1H), 7.32–7.40 (m, 3H), 7.43–7.51 (m, 3H), 7.51–7.56 (m, 2H), 7.62–7.69 (m, 1H), 7.73–7.81 (m, 2H), 8.05 (dd, 1H, J = 7.7, 1.4 Hz); 13C NMR (126 MHz, CDCl3) δ 33.6, 73.6, 76.9, 79.6, 115.8, 121.0, 122.3, 124.6, 126.0, 128.1, 128.8, 129.0, 129.2, 130.0, 130.1, 130.7, 131.3, 136.9, 140.6, 146.0, 165.8, 187.5; IR (cm−1): ν 3197, 2983, 2118, 1716, 1680, 1603, 1468, 1448, 1304, 1039, 870, 760, 752, 694; MS (EI) m/z (%): 419 (13, [M + 1]+), 418 (75, [M]+), 390 (43), 287 (22), 286 (31), 285 (89), 276 (25), 275 (100), 274 (28), 259 (70), 248 (46), 235 (53), 145 (52), 116 (95), 44 (99); HRMS (ESI+): m/z calcd for C26H19N4O2+ [M + H]+ 419.1503, found 419.1502. Anal. Calcd for C26H18N4O2: C, 74.63; H, 4.34; N, 13.39%. Found: C, 74.45; H, 4.40; N, 13.43%.
(1-(3-Methyl-2,4-dioxo-1-(prop-2-yn-1-yl)-1,2,3,4-tetrahydroquinolin-3-yl)-1H-1,2,3-triazol-4-yl)methyl acetate (7c). Colorless crystals, m.p. 159–161 °C (ethyl acetate); Rf = 0.29 (30% ethyl acetate in chloroform); 1H NMR (500 MHz, DMSO-d6) δ 2.06 (s, 3H, COCH3), 2.10 (s, 3H, C-3–CH3), 3.37 (dd, 1H, J = 2.4, 2.4 Hz, C≡CH), 4.84 (dd, 1H, J = 18.1, 2.4 Hz, N-1–CHα), 4.95 (dd, 1H, J = 18.1, 2.4 Hz, N-1–CHβ), 5.17 (s, 2H, OCH2), 7.37 (dd, 1H, J = 7.5, 7.5 Hz, H-6), 7.58 (d, 1H, J = 8.4 Hz, H-8), 7.87–7.93 (m, 1H, H-7), 7.96 (dd, 1H, J = 7.7, 1.5 Hz, H-5), 8.46 (s, 1H, H-5A); 13C NMR (126 MHz, DMSO-d6) δ 20.6 (COCH3), 23.4 (C-3–CH3), 32.6 (N-1–CH2), 57.1 (OCH2), 72.8 (C-3), 75.3 (C≡CH), 78.2 (C≡CH), 116.6 (C-8), 119.2 (C-4a), 124.0 (C-6), 126.0 (C-5A), 128.0 (C-5), 137.1 (C-7), 140.7 (C-8a), 141.5 (C-4A), 167.8 (C-2), 170.1 (COCH3), 189.63 (C-4); 15N NMR (51 MHz, DMSO-d6) δ 134.4 (N1), 247.9 (N-1A), 354.0 (N-3A), 363.4 (N-2A); IR (cm−1): ν 3256, 3152, 2122, 1721, 1687, 1604, 1471, 1383, 1306, 1246, 1194, 1053, 1008, 756; MS (EI) m/z (%): 353 (3, [M + 1]+), 352 (12, [M]+), 213 (69), 212 (34), 184 (19), 156 (32), 146 (17), 130 (19), 129 (21), 128 (22), 77 (17), 57 (16), 55 (23), 43 (100), 42 (17); HRMS (ESI+): m/z calcd for C18H17N4O4+ ([M+H]+): 353.1244, found 353.1246. Anal. Calcd for C18H16N4O4 (352.34): C, 61.36; H, 4.58; N, 15.90%. Found: C, 61.27; H, 4.64; N, 15.87%.
(1-(2,4-Dioxo-3-phenyl-1-(prop-2-yn-1-yl)-1,2,3,4-tetrahydroquinolin-3-yl)-1H-1,2,3-triazol-4-yl)methyl acetate (7d). Colorless crystals, m.p. 210–214 °C; Rf = 0.66 (5% ethanol in chloroform); 1H NMR (500 MHz, DMSO-d6) δ 2.05 (s, 3H, COCH3), 3.41 (dd, 1H, J = 2.4, 2.3 Hz, C≡CH), 4.80 (dd, 1H, J = 18.0, 2.3 Hz, N-1–CHα), 5.09–5.20 (m, 3H, N-1–CHβ, OCH2), 7.24–7.32 (m, 3H, H-6, H-2C, H-6C), 7.41–7.51 (m, 4H, H-8, H-3C, H-4C, H-5C), 7.73–7.79 (m, 1H, H-7), 7.92 (dd, 1H, J = 7.7, 1.5 Hz, H-5), 8.15 (s, 1H, H-5A); 13C NMR (126 MHz, DMSO-d6) δ 20.6 (COCH3), 33.1 (N-1–CH2), 57.1 (OCH2), 75.5 (C≡CH), 77.9 (C≡CH), 80.0 (C-3), 116.3 (C-8), 120.9 (C-4a), 124.2 (C-6), 127.1 (C-5A), 127.8 (C-5), 128.6 (C-2C, C-6C), 129.5 (C-3C, C-5C), 129.9 (C-1C), 130.7 (C-4C), 136.7 (C-7), 140.0 (C-8a), 140.9 (C-4A), 165.8 (C-2), 170.1 (COCH3), 187.7 (C-4); IR (cm−1): ν 3227, 3152, 2116, 1736, 1715, 1683, 1602, 1467, 1379, 1303, 1251, 1036, 764, 747, 694; MS (EI) m/z (%): 415 (2, [M + 1]+), 414 (7, [M]+), 313 (26), 275 (72), 274 (63), 246 (28), 235 (31), 218 (29), 217 (30), 156 (26), 130 (29), 105 (22), 104 (29), 103 (22), 43 (100); HRMS (ESI+): m/z calcd for C23H19N4O4+ [M + H]+ 415.1401, found 415.1403. Anal. Calcd for C23H18N4O4: C, 66.66; H, 4.38; N, 13.52%. Found: C, 66.45; H, 4.39; N, 13.35%.
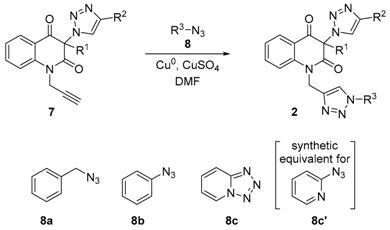
3.8. General Procedure for the Synthesis of Bis-Triazoles 2a,b,d,e,g,h,j,k by Employing CuSO4/Cu0/DMF Conditions (Table 4, Entries 1, 2, 4, 8, 10, 11, 13 and 14)
A solution of azidobenzene (
8b, 197 mg, 1.65 mmol) or (azidomethyl)benzene (
8a, 220 mg, 1.65 mmol) in DMF (4 mL) was added to a vigorously stirred mixture of the appropriate
N‑propargylquinoline-2,4(1
H,3
H)-dione
7 (1.5 mmol), CuSO
4∙5H
2O (38 mg, 0.15 mmol) and granular copper (191 mg, 3.05 mmol) in DMF (5 mL). The reaction mixture was stirred in darkness at room temperature for the time given in
Table 4. The color of the mixture became brown-black. Then, (NH
4)
2CO
3 (432 mg, 4.5 mmol) and water (2 mL) were added to the reaction mixture and the stirring was continued for 10 min. The reaction mixture was poured into a narrow (1 cm in diameter) column of silica gel (15 g). The organic portion was eluted with 10% ethanol in chloroform (approximately 150 mL). The yellow eluate was washed with saturated aqueous NH
4Cl (50 mL), dried over anhydrous sodium sulfate, filtered, and the solvent was removed by rotary evaporation
in vacuo. The TLC pure product thus prepared, with the exception of compounds
2d,
e,
k, was crystallized from suitable solvent. The yields of prepared compounds
2 are given in
Table 4.
1-((1-Benzyl-1H-1,2,3-triazol-4-yl)methyl)-3-methyl-3-(4-phenyl-1H-1,2,3-triazol-1-yl)quinoline-2,4(1H,3H)-dione (2a). Colorless crystals, m.p. 202–204 °C (ethanol); Rf = 0.40 (30% ethyl acetate in chloroform); 1H NMR (500 MHz, DMSO-d6) δ 2.18 (s, 3H, CH3), 5.24 (d, 1H, J = 16.2 Hz, N-1–CHα), 5.49 (d, 1H, J = 16.2 Hz, N-1–CHβ), 5.58 (s, 2H, N-1D–CH2), 7.24–7.29 (m, 2H, H-2E, H-6E), 7.29–7.40 (m, 5H, H-6, H-4B, H-3E, H-4E, H-5E), 7.49 (dd, 2H, J = 7.7, 7.7 Hz, H-3B, H-5B), 7.67 (d, 1H, J = 8.5 Hz, H-8), 7.79–7.90 (m, 3H, H-7, H-2B, H-6B), 7.96 (dd, 1H, J = 7.7, 1.4 Hz, H-5), 8.16 (s, 1H, H-5D), 8.87 (s, 1H, H-5A); 13C NMR (126 MHz, DMSO-d6) δ 23.4 (CH3), 38.7 (N-1–CH2), 52.8 (N-1D–CH2), 72.8 (C-3), 116.7 (C-8), 119.1 (C-4a), 122.5 (C-5A), 123.8 (C-5D), 123.9 (C-6), 125.1 (C-2B, C-6B), 127.9 (C-2E, C-6E), 128.0 (C-4B), 128.1 (C-5), 128.1 (C-4E), 128.7 (C-3E, C-5E), 129.1 (C-3B, C-5B), 130.6 (C-1B), 136.0 (C-1E), 137.2 (C-7), 141.5 (C-8a), 142.2 (C-4D), 145.9 (C-4A), 168.2 (C-2), 190.0 (C-4); IR (cm−1): ν 3137, 3128, 1711, 1673, 1600, 1471, 1387, 1051, 768, 761, 718, 694; MS (EI) m/z (%): 490 (2, [M + 1]+), 489 (6, [M]+), 289 (13), 145 (17), 144 (16), 117 (11), 116 (44), 91 (100), 90 (10), 89 (12); HRMS (ESI+): m/z calcd for C28H24N7O2+ [M + H]+ 490.1986, found 490.1981. Anal. Calcd for C28H23N7O2 (489.53) C, 68.70; H, 4.74; N, 20.03. Found: C, 68.71; H, 4.78; N, 20.36.
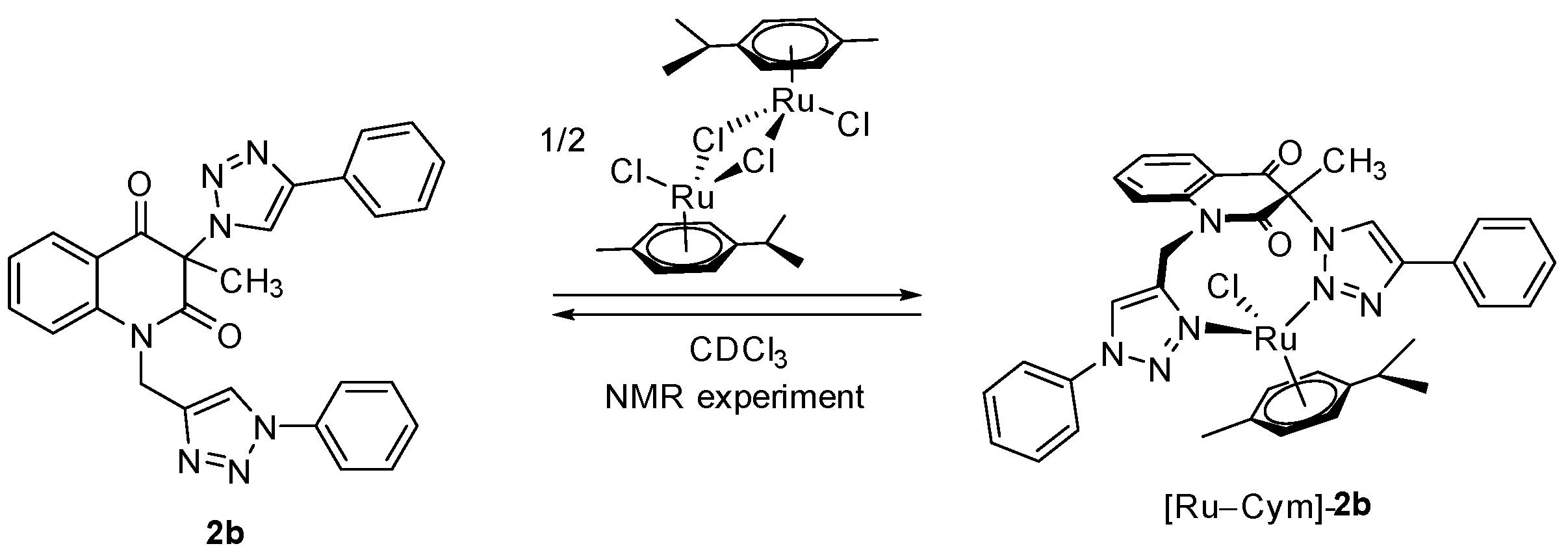
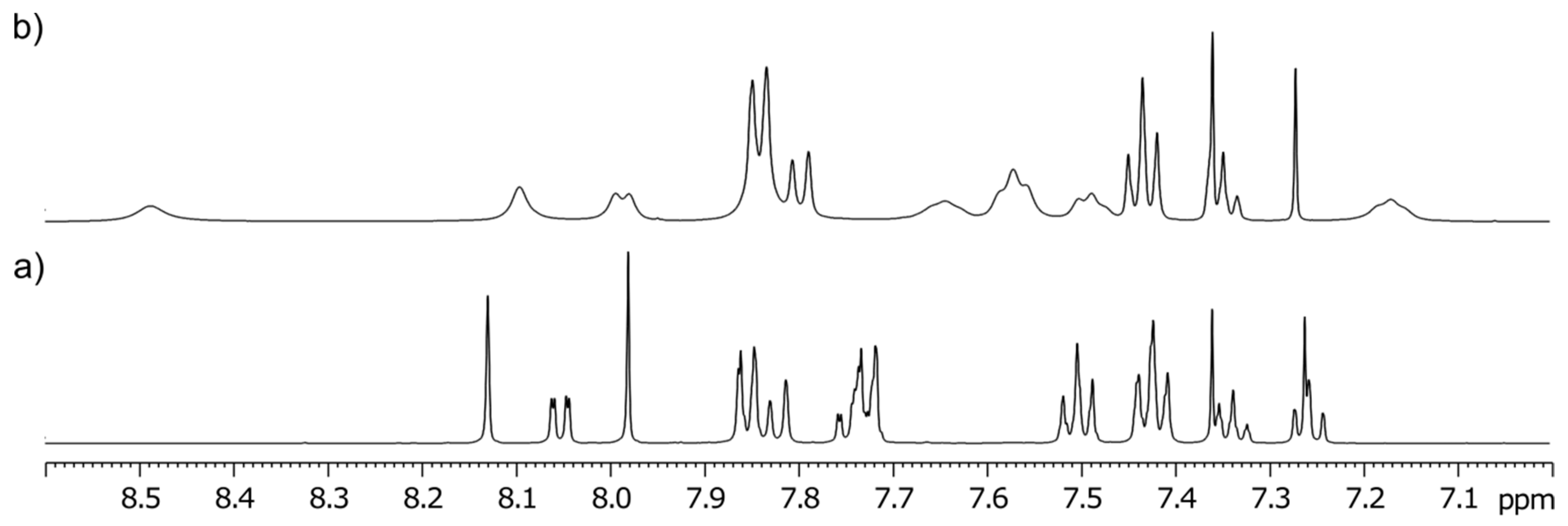
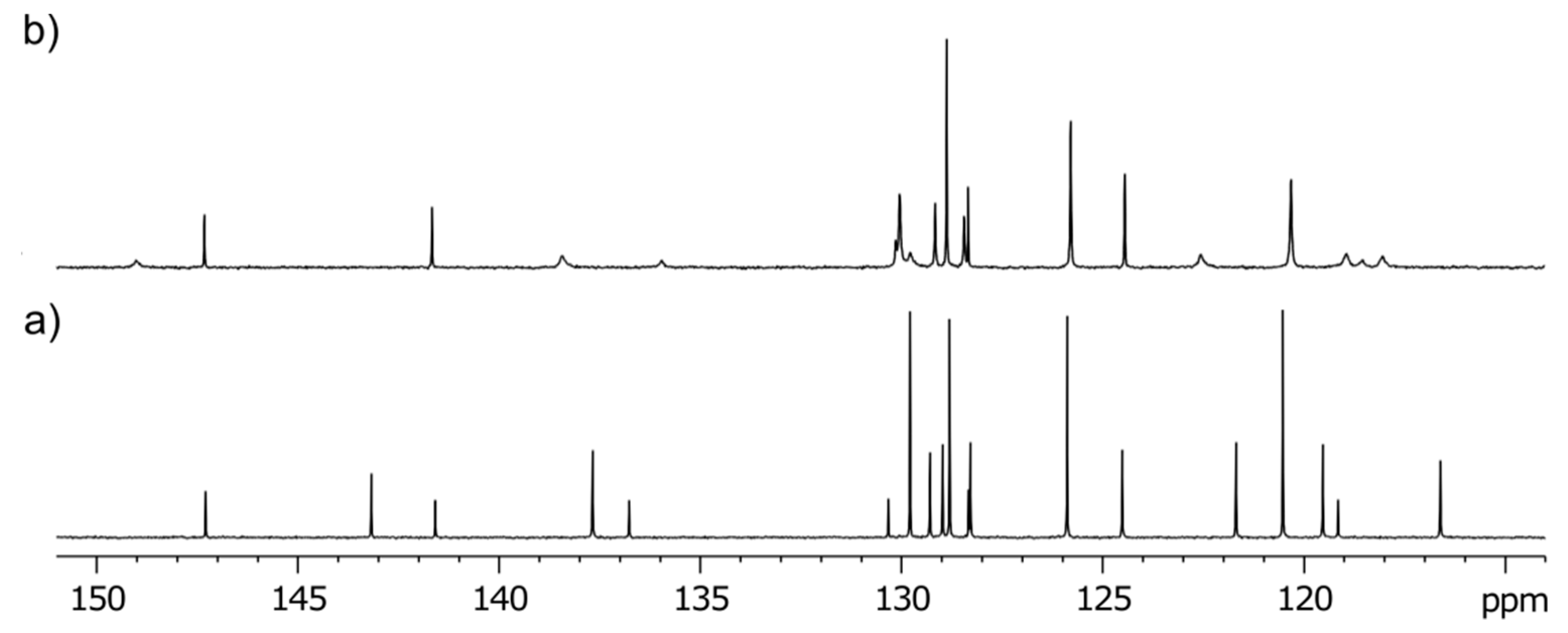
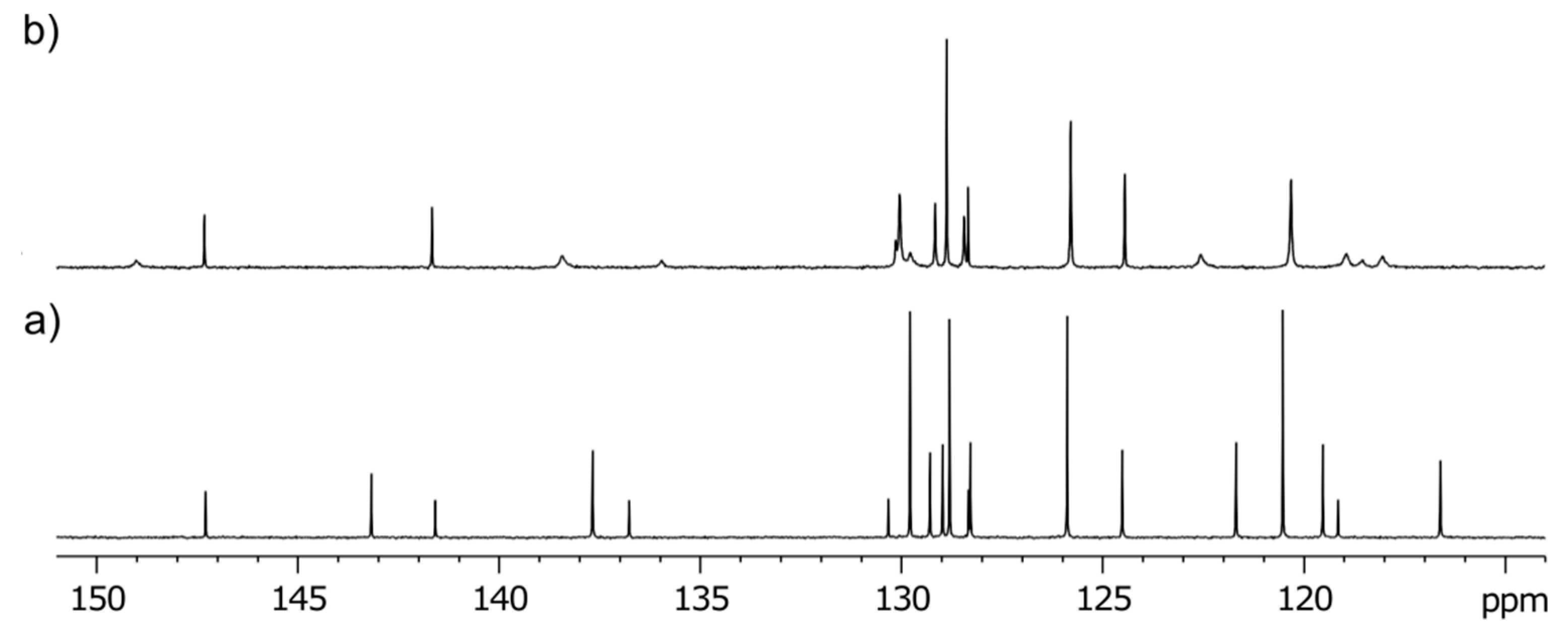
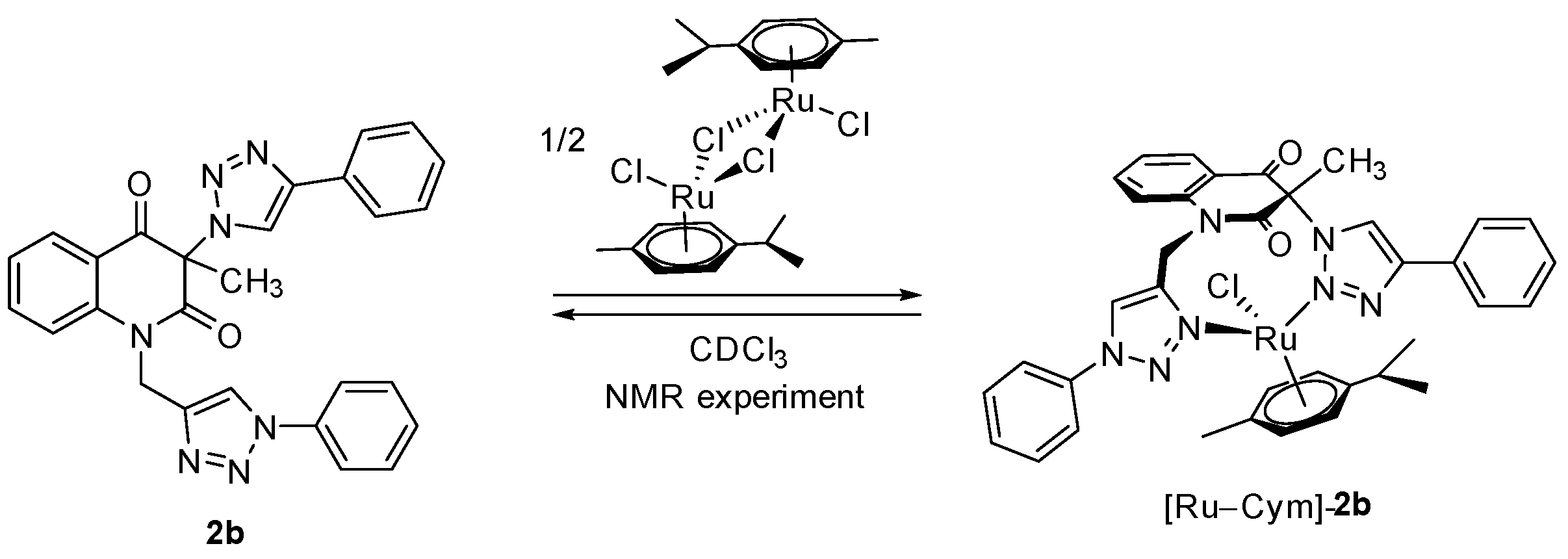
3-Methyl-3-(4-phenyl-1H-1,2,3-triazol-1-yl)-1-((1-phenyl-1H-1,2,3-triazol-4-yl)methyl)quinoline-2,4(1H,3H)-dione (2b). Colorless crystals, m.p. 194–197 °C (benzene); Rf = 0.48 (30% ethyl acetate in chloroform); 1H NMR (500 MHz, DMSO-d6) δ 2.23 (s, 3H, CH3), 5.33 (d, 1H, J = 16.4 Hz, N-1–CHα), 5.62 (d, 1H, J = 16.4 Hz, N-1–CHβ), 7.31–7.39 (m, 2H, H-6, H-4B), 7.45–7.52 (m, 3H, H-3B, H-5B, H-4E), 7.55–7.62 (m, 2H, H-3E, H-5E), 7.70 (d, 1H, J = 8.5 Hz, H-8), 7.82–7.91 (m, 5H, H-7, H‑2B, H-6B, H-2E, H-6E), 7.98 (dd, 1H, J = 7.7, 1.5 Hz, H-5), 8.75 (s, 1H, H-5D), 8.87 (s, 1H, H-5A); 13C NMR (126 MHz, DMSO-d6) δ 23.4 (CH3), 38.7 (N-1–CH2), 73.0 (C-3), 116.8 (C-8), 119.2 (C-4a), 120.2 (C-2E, C-6E), 121.8 (C-5D), 122.5 (C-5A), 124.0 (C-6), 125.2 (C-2B, C-6B), 128.1 (C-4B), 128.1 (C-5), 128.8 (C-4E), 129.1 (C-3B, C-5B), 129.9 (C-3E, C-5E), 130.6 (C-1B), 136.5 (C-1E), 137.3 (C-7), 141.6 (C-8a), 143.3 (C-4D), 146.0 (C-4A), 168.3 (C-2), 190.0 (C-4); 15N NMR (51 MHz, DMSO-d6) δ 136.3 (N1), 248.9 (N-1A), 255.7 (N-D-1), 347.1 (N-3A), 353.4 (N-3D), 358.1 (N-2D), 363.2 (N-2A); IR (cm−1): ν 3275, 1721, 1690, 1613, 1485, 1353, 854, 771, 756, 698, 666, 607, 520; MS (EI) m/z (%): 476 (3, [M + 1]+), 475 (8, [M]+), 289 (14), 145 (12), 131 (11), 130 (100), 129 (18), 128 (11), 116 (56), 104 (12), 103 (16), 102 (12), 89 (12), 77 (69); HRMS (ESI+): m/z calcd for C27H22N7O2+ [M + H]+ 476.1829, found 476.1825. Anal. Calcd for C27H21N7O2 (475.50): C, 68.20; H, 4.45; N, 20.62%. Found: C, 68.48; H, 4.53; N, 20.60%.
(1-(1-((1-Benzyl-1H-1,2,3-triazol-4-yl)methyl)-3-methyl-2,4-dioxo-1,2,3,4-tetrahydroquinolin-3-yl)-1H-1,2,3-triazol-4-yl)methyl acetate (2d). Colorless powder, m.p. 69–82 °C; Rf = 0.42 (30% ethyl acetate in chloroform); 1H NMR (500 MHz, CDCl3), δ 2.09 (s, 3H, COCH3), 2.12 (s, 3H, C-3–CH3), 5.25 (s, 2H, OCH2), 5.33 (s, 2H, N-1-CH2), 5.45 (d, 1H, J = 14.8 Hz, N-1D–CHα), 5.51 (d, 1H, J = 14.8 Hz, N-1D–CHβ), 7.23–7.26 (m, 3H, H-6, H-2E, H-6E), 7.32–7.38 (m, 3H, H-3E, H-4E, H-5E), 7.55 (s, 1H, H-5D), 7.73 (ddd, 1H, J = 8.7, 7.1, 1.6 Hz, H-7), 7.78 (s, 1H, H-5A), 7.82 (d, 1H, J = 8.4 Hz, H-8), 8.02 (dd, 1H, J = 7.7, 1.6 Hz, H-5); 13C NMR (126 MHz, CDCl3) δ 21.1 (COCH3), 23.5 (C-3–CH3), 39.5 (N-1–CH2), 54.5 (N-1D–CH2), 57.7 (OCH2), 71.6 (C-3), 116.9 (C-8), 119.2 (C-4a), 123.5 (C-5D), 124.2 (C-5A), 124.6 (C-6), 128.3 (C-2E, C-6E), 129.0 (C-4E), 129.3 (C-3E, C-5E), 129.3 (C-5), 134.4 (C-1E), 137.8 (C-7), 141.7 (C-8a), 142.3 (C-4A), 142.9 (C-4D), 168.2 (C-2), 171.1 (COCH3), 189.4 (C-4); 15N NMR (51 MHz, CDCl3) δ 138.7 (N-1), 248.4 (N-1A), 250.4 (N-1D), 350.0 (N-3D), 355.2 (N-3A), 361.6 (N-2A), 362.6 (N-2D); IR (cm−1): ν 3143, 2930, 1739, 1717, 1679, 1602, 1470, 1384, 1243, 1186, 1050, 1028, 765, 721, 664; MS (EI) m/z (%): 486 (0.3, [M + 1]+), 485 (1, [M]+), 144 (18), 91 (100), 43 (24); HRMS (ESI+): m/z calcd for C25H24N7O4+ [M + H]+ 486.1884, found 486.1884.
(1-(3-Methyl-2,4-dioxo-1-((1-phenyl-1H-1,2,3-triazol-4-yl)methyl)-1,2,3,4-tetrahydroquinolin-3-yl)-1H-1,2,3-triazol-4-yl)methyl acetate (2e). Colorless powder, m.p. 78–97 °C; Rf = 0.25 (30% ethyl acetate in chloroform); 1H NMR (500 MHz, CDCl3) δ 2.10 (s, 3H, COCH3), 2.20 (s, 3H, C-3–CH3), 5.27 (s, 2H, OCH2), 5.42 (d, 1H, J = 15.8 Hz, N-1–CHα), 5.52 (d, 1H, J = 15.8 Hz, N-1–CHβ), 7.27–7.30 (m, 1H, H-6), 7.41–7.47 (m, 1H, H-4E), 7.49–7.55 (m, 2H, H-3E, H-5E), 7.69–7.74 (m, 2H, H-2E, H-6E), 7.76 (ddd, 1H, J = 8.1, 7.7, 1.6 Hz, H-7), 7.85 (d, 1H, J = 7.3 Hz, H-8), 7.86 (s, 1H, H-5A), 8.05 (dd, 1H, J = 7.8, 1.5 Hz, H-5), 8.10 (s, 1H, H-5D); 13C NMR (126 MHz, CDCl3) δ 21.0 (COCH3), 23.4 (C-3–CH3), 39.5 (N-1-CH2), 57.7 (OCH2), 71.5 (C-3), 116.8 (C-8), 119.2 (C-4a), 120.6 (C-2E, C-6E), 121.7 (C-5D), 124.1 (C-5A), 124.7 (C-6), 129.1 (C-4E), 129.4 (C-5), 129.9 (C-3E, C-5E), 136.9 (C-1E), 137.8 (C-7), 141.7 (C-8a), 142.3 (C-4A), 143.2 (C-4D), 168.3 (C-2), 171.1 (COCH3), 189.4 (C-4); 15N NMR (51 MHz, CDCl3) δ 138.7 (N-1), 248.8 (N-1A), 256.3 (N-1D), 351.9 (N-3D), 355.5 (N-3A); IR (cm−1): ν 3145, 2926, 1740, 1717, 1681, 1601, 1470, 1384, 1242, 1184, 1046, 761, 691, 664; MS (EI) m/z (%): 472 (0.9, [M + 1]+), 471 (3, [M]+), 303 (20), 302 (17), 131 (13), 130 (100), 129 (14), 77 (44), 43 (25); HRMS (ESI+): m/z calcd for C24H22N7O4+ [M + H]+ 472.1728, found 472.1726.
1-((1-Benzyl-1H-1,2,3-triazol-4-yl)methyl)-3-phenyl-3-(4-phenyl-1H-1,2,3-triazol-1-yl)quinoline-2,4(1H,3H)-dione (2g). Colorless crystals, m.p. 142–145 °C (ethanol); Rf = 0.42 (30% ethyl acetate in chloroform); 1H NMR (500 MHz, DMSO-d6) δ 5.15 (d, 1H, J = 15.8 Hz, N-1–CHα), 5.62 (s, 2H, N-1D–CH2), 5.63 (d, 1H, J = 15.8 Hz, N-1–CHβ), 7.22–7.50 (m, 14H, H-6, H-3B, H-4B, H-5B, H-2C, H-3C, H-4C, H-5C, H-6C, H-2E, H-3E, H-4E, H-5E, H-6E), 7.68 (d, 1H, J = 7.8 Hz, H-8), 7.73 (ddd, 1H, J = 8.5, 7.1, 1.7 Hz, H-7), 7.80–7.83 (m, 2H, H-2B, H-6B), 7.92 (dd, 1H, J = 7.7, 1.5 Hz, H-5), 8.24 (s, 1H, H-5D), 8.51 (s, 1H, H-5A); 13C NMR (126 MHz, DMSO-d6) δ 39.2 (N-1–CH2), 52.8 (N-1D–CH2), 80.1 (C-3), 116.7 (C-8), 120.9 (C-4a), 123.4 (C-5A), 124.0 (C-6), 124.2 (C-5D), 125.2 (C-2B, C-6B), 127.9 (C-5), 128.0 (C-4C, C-2E, C-6E), 128.2 (C-4B),128.7 (C-1C), 128.8 (C-3E, C-5E), 129.0 (C-3B, C-5B), 129.4 (C-2C, C-6C), 129.8 (C-1B), 130.5 (C-3C, C-5C, C-4E), 136.0 (C-1E), 136.8 (C-7), 140.8 (C-8a), 141.9 (C-4D), 145.4 (C-4A), 166.2 (C-2), 188.2 (C-4); IR (cm−1): ν 3434, 3138, 3062, 1716, 1678, 1601, 1468, 1375, 1307, 1035, 870, 761, 724, 695; MS (EI) m/z (%): 552 (1, [M + 1]+), 551 (3, [M]+), 289 (23), 236 (11), 145 (18), 144 (17), 116 (31), 104 (10), 91 (100), 89 (11), 77 (16); HRMS (ESI+): m/z calcd for C33H26N7O2+ [M + H]+ 552.2142, found 552.2133. Anal. Calcd for C33H25N7O2 (551.60): C, 71.86; H, 4.57; N, 17.78%. Found: C, 71.58; H, 4.58; N, 17.73%.
3-Phenyl-3-(4-phenyl-1H-1,2,3-triazol-1-yl)-1-((1-phenyl-1H-1,2,3-triazol-4-yl)methyl)quinoline-2,4(1H,3H)-dione (2h). Colorless crystals, m.p. 152–157 °C (ethanol); Rf = 0.54 (30% ethyl acetate in chloroform); 1H NMR (500 MHz, DMSO-d6) δ 5.33 (d, 1H, J = 16.1 Hz, N-1–CHα), 5.71 (d, 1H, J = 16.1 Hz, N-1–CHβ), 7.25–7.29 (m, 1H, H-6), 7.32–7.48 (m, 8H, H-3B, H-4B, H-5B, H-2C, H-3C, H-4C, H-5C, H-6C), 7.48–7.53 (m, 1H, H-4E), 7.58–7.64 (m, 2H, H-3E, H-5E), 7.70 (d, 1H, J = 8.2 Hz, H-8), 7.72–7.77 (m, 1H, H-7), 7.81–7.85 (m, 2H, H-2B, H-6B), 7.88–7.93 (m, 2H, H-2E, H-6E), 7.95 (dd, 1H, J = 7.7, 1.5 Hz, H-5), 8.54 (s, 1H, H-5A), 8.83 (s, 1H, H-5D); 13C NMR (126 MHz, DMSO-d6) δ 39.0 (N-1–CH2), 80.3 (C-3), 116.7 (C-8), 120.2 (C-2E, C-6E), 120.9 (C-4a), 122.3 (C-5D), 123.5 (C-5A), 124.1 (C-6), 125.2 (C-2B, C-6B), 127.9 (C-5), 128.0 (C-4B), 128.9 (C-4E), 128.9 (C-2C, C-6C), 129.0 (C-3B, C-3B), 129.4 (C-3C, C-5C), 129.9 (C-1C), 130.0 (C-3E, C-5E), 130.5 (C-4C), 130.6 (C-1B), 136.5 (C-1E), 136.8 (C-7), 140.7 (C-8a), 142.9 (C-4D), 145.4 (C-4A), 166.4 (C-2), 188.2 (C-4); IR (cm−1): ν 3447, 3142, 3060, 1716, 1679, 1600, 1468, 1449, 1375, 1305, 1040, 871, 758, 693; MS (EI) m/z (%): 538 (1, [M + 1]+), 537 (3, [M]+), 366 (14), 262 (10), 236 (17), 145 (29), 131 (10), 130 (100), 129 (19), 128 (11), 118 (10), 116 (38), 104 (14), 103 (17), 102 (13), 90 (12), 89 (15), 77 (71), 51 (12); HRMS (ESI+): m/z calcd for C32H24N7O2+ ([M+H]+) 538.1986, found 538.1976. Anal. Calcd for Anal. calcd for C32H23N7O2 (537.57) C, 71.50; H, 4.31; N, 18.24%. Found: C, 71.22; H, 4.32; N, 17.94%.
(1-(1-((1-Benzyl-1H-1,2,3-triazol-4-yl)methyl)-2,4-dioxo-3-phenyl-1,2,3,4-tetrahydroquinolin-3-yl)-1H-1,2,3-triazol-4-yl)methyl acetate (2j). Colorless powder, m.p. 188–194 °C (ethanol); Rf = 0.41 (30% ethyl acetate in chloroform); 1H NMR (500 MHz, CDCl3) δ 2.04 (s, 3H, CH3), 5.17 (s, 2H, OCH2), 5.21 (d, 1H, J = 15.6 Hz, N-1–CHα), 5.43 (d, 1H, J = 14.8 Hz, N-1D–CHα), 5.51 (d, 1H, J = 15.6 Hz, N-1–CHβ), 5.55 (d, 1H, J = 14.8 Hz, N-1D–CHβ), 7.08 (s, 1H, H-5A), 7.18 (ddd, 1H, J = 7.5, 7.5, 0.8 Hz, H-6), 7.23–7.29 (m, 4H, H-3C, H-5C, H-2E, H-6E), 7.29–7.33 (m, 2H, H-2C, H-6C), 7.34–7.39 (m, 3H, H-3E, H-4E, H-5E), 7.38–7.44 (m, 1H, H-4C), 7.58 (s, 1H, H-5D), 7.63 (ddd, 1H, J = 8.4, 7.4, 1.7 Hz, H-7), 7.75 (d, 1H, J = 8.3 Hz, H-8), 7.99 (dd, 1H, J = 7.7, 1.7 Hz, H-5); 13C NMR (126 MHz, CDCl3) δ 21.0 (CH3), 39.9 (N-1–CH2), 54.5 (N-1D–CH2), 57.6 (OCH2), 79.6 (C-3), 116.8 (C-8), 120.9 (C-4a), 123.5 (C-5D), 124.6 (C-6), 126.4 (C-5A), 128.3 (C-2E, C-6E), 128.7 (C-2C, C-6C), 129.0 (C-5), 129.1 (C-4E), 129.4 (C-3E, C-5E), 129.7 (C-1C), 130.0 (C-3C, C-5C), 131.3 (C-4C), 134.5 (C-1E), 137.2 (C-7), 140.9 (C-4A), 141.1 (C-8a), 142.9 (C-4D), 166.6 (C-2), 171.0 (COCH3), 187.9 (C-4); 15N NMR (51 MHz, CDCl3) δ 140.4 (N-1), 249.8 (N-1A), 250.4 (N-1D), 350.5 (N-3D), 356.9 (N-3A), 362.9 (N-2D), 365.1 (N-2A); IR (cm−1): ν 3142, 2927, 1740, 1717, 1679, 1602, 1469, 1377, 1244, 768, 749, 714, 697; MS (EI) m/z (%): 548 (0.1, [M + 1]+), 547 (0.3, [M]+), 347 (13), 289 (13), 144 (14), 105 (10), 104 (13), 91 (100), 43 (29); HRMS (ESI+): m/z calcd for C30H26N7O4+ [M + H]+ 548.2041, found 548.2032.
(1-(2,4-Dioxo-3-phenyl-1-((1-phenyl-1H-1,2,3-triazol-4-yl)methyl)-1,2,3,4-tetrahydroquinolin-3-yl)-1H-1,2,3-triazol-4-yl)methyl acetate (2k). Colorless powder, m.p. 93–105 °C; Rf = 0.42 (30% ethyl acetate in chloroform); 1H NMR (500 MHz, CDCl3) δ 2.05 (s, 3H, CH3), 5.19 (s, 2H, OCH2), 5.42 (d, 1H, J = 15.7 Hz, N-1–CHα), 5.55 (d, 1H, J = 15.7 Hz, N-1–CHβ), 7.14 (s, 1H, H-5A), 7.20 (ddd, 1H, J = 7.6, 7.6, 0.8 Hz, H-6), 7.38–7.49 (m, 6H, H-2C, H-3C, H-4C, H-5C, H-6C, H-4E), 7.49–7.55 (m, 2H, H-3E, H-5E), 7.66 (ddd, 1H, J = 8.5, 7.3, 1.7 Hz, H-7), 7.68–7.72 (m, 2H, H-2E, H-6E), 7.76 (d, 1H, J = 8.4 Hz, H-8), 8.03 (dd, 1H, J = 7.8, 1.5 Hz, H-5), 8.05 (s, 1H, H-5D); 13C NMR (126 MHz, CDCl3) δ 21.0 (CH3), 39.8 (N-1-CH2), 57.6 (OCH2), 79.6 (C-3), 116.7 (C-8), 120.7 (C-2E, C-6E), 120.9 (C-4a), 121.8 (C-5D), 124.7 (C-6), 126.4 (C-5A), 128.9 (C-2C, C-6C), 129.1 (C-5), 129.2 (C-4E), 129.9 (C-1C), 130.0 (C-3E, C-5E), 130.2 (C-3C, C-5C), 131.4 (C-4C), 136.9 (C-1E), 137.4 (C-7), 140.9 (C-4A), 140.9 (C-8a), 143.2 (C-4D), 166.9 (C-2), 171.0 (COCH3), 187.9 (C-4); 15N NMR (51 MHz, CDCl3) δ 140.4 (N-1), 249.9 (N-1A), 256.3 (N-1D), 352.9 (N-3D), 357.2 (N-3A); IR (cm−1): ν 3146, 2962, 1741, 1718, 1681, 1600, 1468, 1376, 1243, 1043, 762, 693, 665, 608; MS (EI) m/z (%): 534 (0.2, [M + 1]+), 533 (0.6, [M]+), 366 (12), 365 (11), 262 (12), 131 (11), 130 (100), 129 (19), 128 (12), 104 (14), 103 (16), 99 (18), 77 (62), 44 (17), 43 (52); HRMS (ESI+): m/z calcd for C29H24N7O4+ [M + H]+ 534.1884, found 534.1882.
3.9. General Procedure for the Synthesis of Bis-Triazoles 2c,f,i,l by Employing CuSO4/Cu0/DMF Conditions (Table 4, Entries 3, 9, 12 and 15)
A mixture of the appropriate
N-propargylquinoline-2,4(1
H,3
H)-dione
7 (1.5 mmol), tetrazolo[1,5-
a]pyridine (189 mg, 1.58 mmol), CuSO
4∙5H
2O (38 mg, 0.15 mmol), granular copper (191 mg, 3.05 mmol) and DMF (9 mL) was heated in darkness to 95–105 °C (oil bath) for the time given in
Table 4, whereas the color of the mixture changed from brown-black to dark green. The mixture was then allowed to cool to room temperature. Subsequently, (NH
4)
2CO
3 (432 mg, 4.5 mmol) and water (2 mL) were added and after stirring for 15 min, the mixture was poured into a narrow (1 cm diameter) column of silica gel (15 g). The organic portion was eluted from the column with 10% ethanol in chloroform. The yellow eluate was washed with saturated aqueous NH
4Cl (50 mL), dried over anhydrous sodium sulfate, filtered, and the solvent was removed by rotary evaporation
in vacuo. In the cases of
2c,
i, the residue, which was TLC pure compound, was crystallized from suitable solvent. In the cases of
2f,
l, the residue was purified by chromatography on silica gel column using chloroform as eluent. The yields of prepared compounds
2 are given in
Table 4.
3-Methyl-3-(4-phenyl-1H-1,2,3-triazol-1-yl)-1-((1-(pyridin-2-yl)-1H-1,2,3-triazol-4-yl)methyl)quinoline-2,4(1H,3H)-dione (2c). Colorless crystals, m.p. 188–191 °C (benzene); Rf = 0.29 (30% ethyl acetate in chloroform); 1H NMR (500 MHz, DMSO-d6) δ 2.23 (s, 3H, CH3), 5.42 (d, 1H, J = 16.5 Hz, N-1–CHα), 5.58 (d, 1H, J = 16.5 Hz, N-1–CHβ), 7.28–7.40 (m, 2H, H-6, H-4B), 7.43–7.51 (m, 2H, H-3B, H-5B), 7.51–7.57 (m, 1H, H-5E), 7.64 (d, 1H, J = 8.4 Hz, H-8), 7.78–7.90 (m, 3H, H-7, H-2B, H-6B), 7.99 (d, 1H, J = 7.5 Hz, H-5), 8.07–8.17 (m, 2H, H-3E, H-4E), 8.54–8.61 (m, 1H, H-6E), 8.82 (s, 1H, H-5D), 8.87 (s, 1H, H-5A); 13C NMR (126 MHz, DMSO-d6) δ 23.4 (CH3), 38.7 (N-1–CH2), 73.0 (C-3), 113.7 (C-3E), 116.6 (C-8), 119.3 (C-4a), 120.6 (C-5D), 122.5 (C-5A), 123.9 (C-6), 124.5 (C-5E), 125.2 (C-2B, C-6B), 128.1 (C-4B), 128.1 (C-5), 129.1 (C-3B, C-5B), 130.5 (C-1B), 137.2 (C-7), 140.3 (C-4E), 141.4 (C-8a), 143.2 (C-4D), 146.0 (C-4A), 148.4 (C-2E), 149.0 (C-6E), 168.5 (C-2), 189.9 (C-4); 15N NMR (51 MHz, DMSO-d6) δ 135.8 (N-1), 248.9 (N-1A), 260.5 (N-1D), 284.9 (N-1E), 347.1 (N-3A), 356.9 (N-3D), 358.6 (N-2D), 363.4 (N-2A); IR (cm−1): ν 3426, 3126, 2972, 1706, 1674, 1601, 1471, 1378, 1310, 1232, 1041, 777, 764; MS (EI) m/z (%): 477 (2, [M + 1]+), 476 (7, [M]+), 289 (11), 145 (14), 132 (14), 131 (96), 116 (50), 102 (10), 90 (10), 89 (13), 79 (20), 78 (100), 77 (10), 51 (10); HRMS (ESI+): m/z calcd for C26H21N8O2+ [M + H]+ 477.1782, found 477.1773. Anal. Calcd for C26H20N8O2 (476.48) C, 65.54; H, 4.23; N, 23.52%. Found: C, 65.68; H, 4.21; N, 23.63%.
(1-(3-Methyl-2,4-dioxo-1-((1-(pyridin-2-yl)-1H-1,2,3-triazol-4-yl)methyl)-1,2,3,4-tetrahydroquinolin-3-yl)-1H-1,2,3-triazol-4-yl)methyl acetate (2f). Colorless powder, m.p. 69–82 °C; Rf = 0.29 (30% ethyl acetate in chloroform); 1H NMR (500 MHz, DMSO-d6) δ 2.06 (s, 3H, COCH3), 2.18 (s, 3H, C3–CH3), 5.17 (d, 1H, J = 12.7 Hz, O–CHα), 5.20 (d, 1H, J = 12.7 Hz, O–CHβ), 5.41 (d, 1H, J = 16.5 Hz, N-1–CHα), 5.53 (d, 1H, J = 16.5 Hz, N-1–CHβ), 7.31 (dd, 1H, J = 7.4, 7.4 Hz, H-6), 7.54 (dd, 1H, J = 8.8, 4.5 Hz, H-5E), 7.59 (d, 1H, J = 8.5 Hz, H-8), 7.77–7.83 (m, 1H, H-7), 7.96 (dd, 1H, J = 7.7 Hz, J = 1.6 Hz, H-5), 8.08–8.14 (m, 2H, H-3E, H-4E), 8.47 (s, 1H, H-5A), 8.55–8.59 (m, 1H, H-6E), 8.82 (s, 1H, H-5D); 13C NMR (126 MHz, DMSO-d6) δ 20.6 (COCH3), 23.5 (C3–CH3), 38.7 (N-1–CH2), 57.2 (OCH2), 73.3 (C-3), 113.7 (C-3E), 116.5 (C-8), 119.4 (C-4a), 120.6 (C-5D), 123.8 (C-6), 124.4 (C-5E), 126.1 (C-5A), 127.9 (C-5), 137.0 (C-7), 140.2 (C-4E), 141.3 (C-8a), 141.6 (C-4A), 143.2 (C-4D), 148.3 (C-2E), 148.9 (C-6E), 168.6 (C-2), 170.1 (COCH3), 189.9 (C-4); 15N NMR (51 MHz, DMSO-d6) δ 135.3 (N1), 247.6 (N-1A), 260.0 (N-1D), 284.7 (N-1E), 353.4 (N-3A), 356.5 (N-3D), 361.9 (N-2D), 363.7 (N-2A); IR (cm−1): ν 3152, 1741, 1718 1681, 1600, 1471, 1384, 1314, 1242, 1183, 1038, 782, 756, 663; MS (EI) m/z (%): 473 (0.7, [M + 1]+), 472 (2, [M]+), 304 (27), 303 (26), 302 (17), 132 (13), 131 (100), 79 (22), 78 (100), 43 (21); HRMS (ESI+): m/z calcd for C23H21N8O4+ [M + H]+ 473.1680, found 473.1684. Anal. Calcd for C23H20N8O4·½H2O (472.46): C, 57.38; H, 4.40; N, 23.27%. Found: C, 57.39; H, 4.36; N, 23.47%.
3-Phenyl-3-(4-phenyl-1H-1,2,3-triazol-1-yl)-1-((1-(pyridin-2-yl)-1H-1,2,3-triazol-4-yl)methyl)quinoline-2,4(1H,3H)-dione (2i). Colorless crystals, m.p. 188–192 °C (benzene); Rf = 0.50 (30% ethyl acetate in chloroform); 1H NMR (500 MHz, DMSO-d6) δ 5.44 (d, 1H, J = 16.3 Hz, N-1–CHα), 5.67 (d, 1H, J = 16.3 Hz, N-1–CHβ), 7.26 (dd, 1H, J = 7.5, 7.5 Hz, H-6), 7.32–7.40 (m, 3H, H-4B, H-2C, H-6C), 7.41–7.52 (m, 5H, H-3B, H-5B, H-3C, H-4C, H-5C), 7.52–7.61 (m, 2H, H-8, H-5E), 7.68–7.75 (m, 1H, H-7), 7.78–7.86 (m, 2H, H-2B, H-6B), 7.96 (dd, 1H, J = 7.7, 1.5 Hz, H-5), 8.09–8.16 (m, 2H, H-3E, H-4E), 8.58 (s, 1H, H-5A), 8.60 (ddd, 1H, J = 4.8, 1.3, 1.3 Hz, H-6E), 8.81 (s, 1H, H-5D); 13C NMR (126 MHz, DMSO-d6) δ 39.1 (N-1–CH2), 80.4 (C-3), 113.7 (C-3E), 116.5 (C-8), 120.8 (C-5D), 120.9 (C-4a), 123.4 (C-5A), 124.0 (C-6), 124.5 (C-5E), 125.2 (C-2B, C-6B), 127.9 (C-5), 128.0 (C-4B), 128.9 (C-2C, C-6C), 129.0 (C-3B, C-5B), 129.4 (C-3C, C-5C), 130.0 (C-1C), 130.5 (C-4C), 130.6 (C-1B), 136.8 (C-7), 140.2 (C-4E), 140.5 (C-8a), 143.0 (C-4D), 145.4 (C-4A), 148.3 (C-2E), 149.0 (C-6E), 166.6 (C-2), 188.1 (C-4); 15N NMR (51 MHz, DMSO-d6) δ 137.5 (N1), 248.7 (N-1A), 260.4 (N-1D), 284.8 (N-1E), 347.2 (N-3A), 357.7 (N-3D), 367.4 (N-2A); IR (cm−1): ν 3418, 2973, 1718, 1679, 1596, 1477, 1467, 1450, 1049, 1031, 773, 766, 757, 701; MS (EI) m/z (%): 539 (1, [M + 1]+), 538 (3, [M]+), 236 (11), 145 (16), 132 (14), 131 (100), 116 (32), 91 (11), 89 (11), 79 (15), 78 (85), 77 (13); HRMS (ESI+): m/z calcd for C31H23N8O2+ [M + H]+ 539.1938, found 539.1932. Anal. calcd for C31H22N8O2 (538.19): C, 69.13; H, 4.12; N, 20.81%. Found: C, 68.91; H, 4.17; N, 20.66%.
(1-(2,4-Dioxo-3-phenyl-1-((1-(pyridin-2-yl)-1H-1,2,3-triazol-4-yl)methyl)-1,2,3,4-tetrahydroquinolin-3-yl)-1H-1,2,3-triazol-4-yl)methyl acetate (2l). Colorless powder, m.p. 93–102 °C; Rf = 0.18 (30% ethyl acetate in chloroform); 1H NMR (500 MHz, CDCl3) δ 2.05 (s, 3H, COCH3), 5.19 (s, 2H, OCH2), 5.30 (d, 1H, J = 15.8 Hz, N-1–CHα), 5.71 (d, 1H, J = 15.8 Hz, N-1–CHβ), 7.13 (s, 1H, H-5A), 7.19 (dd, 1H, J = 7.5, 7.5 Hz, H-6), 7.36 (dd, 1H, J = 7.3, 4.9 Hz, H-5E), 7.38–7.42 (m, 2H, H-3C, H-5C), 7.42–7.48 (m, 3H, H-2C, H-4C, H-6C), 7.63 (ddd, 1H, J = 8.3, 7.4, 1.6 Hz, H-7), 7.70 (d, 1H, J = 8.4 Hz, H-8), 7.88–7.95 (m, 1H, H-4E), 8.02 (dd, 1H, J = 7.8, 1.5 Hz, H-5), 8.15 (d, 1H, J = 8.2 Hz, H-3E), 8.47–8.53 (m, 1H, H-6E), 8.63 (s, 1H, H-5D); 13C NMR (126 MHz, CDCl3) δ 21.0 (COCH3), 39.9 (N-1–CH2), 57.6 (OCH2), 79.7 (C-3), 113.9 (C-3E), 116.6 (C-8), 121.0 (C-4a), 121.0 (C-5D), 124.0 (C-5E), 124.6 (C-6), 126.4 (C-5A), 128.9 (C-2C, C-6C), 129.1 (C-5), 129.7 (C-1C), 130.2 (C-3C, C-5C), 131.3 (C-4C), 137.2 (C-7), 139.3 (C-4E), 140.9 (C-4A), 141.2 (C-8a), 143.0 (C-4D), 148.9 (C-6E), 149.0 (C-2E), 166.6 (C-2), 171.0 (COCH3), 187.9 (C-4); 15N NMR (51 MHz, CDCl3) δ 138.9 (N-1), 249.7 (N-1A), 261.2 (N-1D), 285.1 (N-1E), 355.8 (N-3D), 357.1 (N-3A); IR (cm−1): ν 3155, 2926, 1741, 1718, 1682, 1599, 1470, 1375, 1313, 1243, 1034, 779, 697, 665; MS (EI) m/z (%): 535 (0.4, [M + 1]+), 534 (0.7, [M]+), 132 (14), 131 (100), 79 (19), 78 (93), 44 (11), 43 (31); HRMS (ESI+): m/z calcd for C28H23N8O4+ [M + H]+ 535.1837, found 535.1846.
3.10. Synthesis of Bis-Triazole 2d by Employing CH2Cl2/Water/CuSO4∙5H2O/Na-Ascorbate Conditions (Table 4, Entry 5)
To a solution of acetylene 7c (132 mg, 0.375 mmol) and azide 8a (52.4 mg, 0.394 mmol) in dichloromethane (6.5 mL) a solution of sodium ascorbate (59.5 mg, 0.3 mmol) in water (5.5 mL), and a solution of CuSO4∙5H2O (7.5 mg, 0.03 mmol) in water (1 mL) were added. The two-phase liquid reaction mixture was stirred in darkness at room temperature until the compound 7c reacted completely according to TLC analysis (4 h). The reaction mixture was diluted with water (50 mL) and extracted with chloroform (4 × 30 mL). The combined organic layers were dried (Na2SO4), filtered, and evaporated to dryness. The residue was dissolved in chloroform (5 mL) and subjected to silica gel (25 g) column chromatography using 67% ethyl acetate in petroleum ether as eluent, affording product 2d (155 mg, 0.32 mmol, 85%).
3.11. Synthesis of Bis-Triazole 2d by Employing t-BuOH/Water/CH3CN/CuSO4∙5H2O/Na-Ascorbate Conditions (Table 4, Entry 6)
To a mixture of acetylene 7c (264 mg, 0.75 mmol), azide 8a (105 mg, 0.79 mmol) and t-BuOH (3.5 mL) a solution of Na-ascorbate (30 mg, 0.15 mmol) in water (2.5 mL), and a solution of CuSO4∙5H2O (4 mg, 0.02 mmol) in water (1 mL) were added. The reaction mixture was stirred in darkness at room temperature for 9 h. Then a solution of Na-ascorbate (89 mg, 0.45 mmol) in water (1 mL), and a solution of CuSO4∙5H2O (11 mg, 0.044 mmol) in water (1 mL) and t-BuOH (2 mL) were added. The reaction mixture was stirred for additional 20 h. The resulting sticky sediment that formed in the course of the reaction was dissolved by addition of acetonitrile (3 mL) to the reaction mixture. The reaction mixture was stirred for additional 19 h. Although the azide and acetylene coupling partners were still present in the reaction mixture, as judged by TLC analysis, the reaction was stopped by the addition of water (50 mL) and extracted with chloroform (4 × 30 mL). The combined organic layers were dried (Na2SO4), filtered, and evaporated to dryness. The residue was dissolved in chloroform and subjected to silica gel (35 g) column chromatography using 67% ethyl acetate in petroleum ether as eluent, affording regenerated starting acetylene 7c (48 mg, 0.14 mmol, 18%) and product 2d (295 mg, 0.61 mmol, 81%).
3.12. Synthesis of Bis-Triazole 2d by Employing t-BuOH/Water/CuSO4∙5H2O/l-Ascorbic Acid Conditions (Table 4, Entry 7)
To a mixture of acetylene 7c (264 mg, 0.75 mmol) and azide 8a (105 mg, 0.79 mmol) a solution of l-ascorbic acid (13 mg, 0.074 mmol) and CuSO4∙5H2O (2 mg, 0.008 mmol) in water (3.5 mL), and t-BuOH (3.5 mL) were added. The reaction mixture was stirred in darkness at room temperature. After 8.5 h and 22 h of stirring additional portions of l-ascorbic acid/CuSO4∙5H2O/water/t-BuOH (40 mg, 0.23 mmol/6 mg, 0.02 mmol/1 mL/1 mL and 53 mg, 0.3 mmol/7.5 mg, 0.03 mmol/1 mL/1 mL, respectively) were added. Although after stirring for additional 23 h (total reaction time 45 h), TLC analysis indicated the presence of azide and acetylene starting compounds, the heterogeneous reaction mixture (a sticky sediment was formed) was diluted with water and extracted with chloroform (5 × 50 mL). The combined organic layers were dried (Na2SO4), filtered, and evaporated to dryness. The residue was dissolved in chloroform (5 mL) and subjected to silica gel (35 g) column chromatography using 33% ethyl acetate in petroleum ether as eluent, affording regenerated starting acetylene 7c (114 mg, 0.32 mmol, 43%) and product 2d (165 mg, 0.34 mmol 45%).
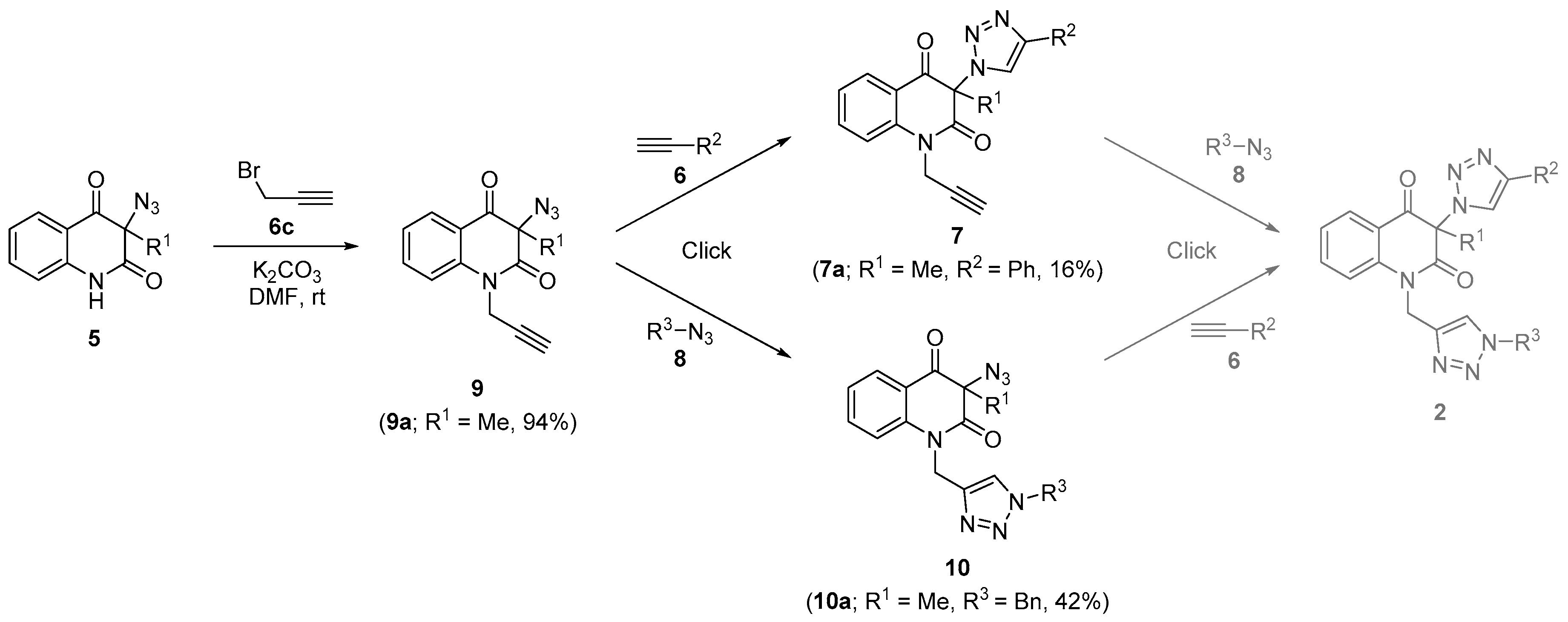
3.13. 3-Azido-3-methyl-1-(prop-2-yn-1-yl)Quinoline-2,4(1H,3H)-Dione (9a) (Scheme 3)
A mixture of the azide 5a (649 mg, 3.0 mmol) and potassium carbonate (1.24 g, 9 mmol) in DMF (15 mL) was stirred at room temperature in darkness for 40 min. Propargyl bromide (6c, 80% solution in toluene, 669 mg, 4.5 mmol) diluted with DMF (7 mL) was added dropwise under stirring during 1 min. The reaction mixture was stirred for 6 h, during which time it turned yellow, diluted with cold water (200 mL) and extracted with chloroform (5 × 50 mL). The combined organic layers were dried (Na2SO4), filtered, and evaporated to dryness. Trace amounts of DMF were removed by five subsequent co-destilations in vacuo at 50 °C with toluene (30 mL). The residual yellow oil was dissolved in chloroform (5 mL) and chromatographed on column silica gel (35 g) using chloroform as eluent, affording product 9a (717 mg, 2.82 mmol, 94%, dried in vacuo to constant weight) as off-white oily material, that was pure by TLC (Rf = 0.57; chloroform); 1H NMR (500 MHz, CDCl3) δ 1.79 (s, 3H), 2.29 (dd, 1H, J = 2.5, 2.5 Hz), 4.67 (dd, 1H, J = 17.8, 2.5 Hz), 4.98 (dd, 1H, J = 17.8, 2.5 Hz), 7.26 (ddd, 1H, J = 7.7, 7.4, 0.8 Hz), 7.35 (d, 1H, J = 8.3 Hz), 7.71 (ddd, 1H, J = 8.3, 7.4, 1.7 Hz), 8.02 (dd, 1H, J = 7.7, 1.7 Hz); 13C NMR (126 MHz, CDCl3) δ 23.6, 32.7, 70.7, 73.4, 77.1, 115.8, 119.6, 124.4, 129.0, 136.9, 140.8, 169.1, 191.1; IR (cm−1): ν 3241, 2980, 2138, 2107, 1711, 1678, 1603, 1471, 1383, 1366, 1305, 1285, 1260, 1218, 762; HRMS (ESI+): m/z calcd for C13H11N4O2+ [M + H]+ 255.0877, found 255.0877; calcd for C13H11N2O2+ [M − N2 + H]+ 227.0815, found 227.0814.
3.14. Synthesis of Triazole 7a from Phenylacetylene (6a) and Compound 9a (Scheme 3)
A mixture of compound 9a (286 mg, 1.13 mmol), phenylacetylene (6a) (230 mg, 2.25 mmol), CuSO4 5H2O (28 mg, 0.11 mmol) and granular copper (143 mg, 2.25 mmol) in DMF (5 mL) was stirred in darkness at room temperature for 60 min. To the resulting brown-green suspension (NH4)2CO3 (324 mg, 3.38 mmol) and water (3 mL) were added and stirring was continued for 10 min. The resulting mixture was diluted with 10% ethanol in chloroform (10 mL). The organic layer was separated and the aqueous layer was extracted with chloroform (3 × 10 mL). The combined organic layers were passed through a narrow (1 cm in diameter) column of silica gel (13 g) and the column was subsequently washed with 10% ethanol in chloroform (210 mL) using overpressure to the top of the column. The yellow eluate was washed with saturated aqueous NH4Cl (1 × 50 mL) and distilled water (1 × 50 mL), dried (Na2SO4), filtered, and evaporated to dryness. Trace amounts of DMF were removed by five subsequent co-destilations in vacuo at 50 °C with toluene (40 mL). The residue was chromatographed on a column of silica gel (30 g) using 38% ethyl acetate in hexane. The resulting white solid (88 mg) was crystallized from benzene affording triazole 7a (66 mg, 0.19 mmol, 16%), which was identified with the compound 7a described above.
3.15. 3-Azido-1-((1-Benzyl-1H-1,2,3-Triazol-4-yl)Methyl)-3-Methylquinoline-2,4(1H,3H)-Dione (10a) (Scheme 3)
A mixture of acetylene 9a (254 mg, 1.0 mmol), (azidomethyl)benzene (8a) (266 mg, 2.0 mmol), CuSO4∙5H2O (25 mg, 0.1 mmol) and granular copper (127 mg, 2.0 mmol) in DMF (10 mL) was stirred at room temperature for 21 h. Then (NH4)2CO3 (288 mg, 3.0 mmol) and water (3 mL) were added and the stirring was continued for 10 min. The resulting mixture was poured into a narrow (1 cm in diameter) column of silica gel (13 g). The organic portion was eluted with 10% ethanol in chloroform (190 mL). The yellow eluate was washed with saturated aqueous NH4Cl (50 mL) and water (50 mL), dried (Na2SO4), filtered, and evaporated to dryness. Trace amounts of DMF were removed by six subsequent co-destilations in vacuo at 50 °C with toluene (30 mL). The residue was dissolved in chloroform (5 mL) and chromatographed on silica gel (35 g) column using gradually 38% and 50% ethyl acetate in petroleum ether as mobile phase, affording product 10a (164 mg, 0.42 mmol, 42%) as a white solid, m.p. 42–47 °C; Rf = 0.21 (38% ethyl acetate in petroleum ether); 1H NMR (500 MHz, CDCl3) δ 1.73 (s, 3H), 5.20 (d, 1H, J = 15.6 Hz), 5.31 (d, 1H, J = 15.6 Hz), 5.45 (d, 1H, J = 14.8 Hz), 5.49 (d, 1H, J = 14.8 Hz), 7.16–7.28 (m, 3H), 7.32–7.39 (m, 3H), 7.54 (s, 1H), 7.67 (ddd, 1H, J = 8.3, 7.4, 1.6 Hz), 7.77 (d, 1H, J = 8.4 Hz), 7.96 (dd, 1H, J = 7.7, 1.5 Hz); 13C NMR (126 MHz, CDCl3) δ 23.6, 39.1, 54.5, 70.6, 116.6, 119.5, 123.5, 124.3, 128.3, 128.8, 129.0, 129.3, 134.3, 137.1, 141.4, 143.0, 169.8, 191.3; IR (cm−1): ν 3137, 3033, 2980, 2106, 1713, 1676, 1602, 1489, 1469, 1379, 1336, 1279, 1223, 765, 724; HRMS (ESI+): m/z calcd for C20H18N7O2+ [M + H]+ 388,1516, found 388.1514. Anal. Calcd for C20H17N7O2 (387.39): C, 62.01; H, 4.42; N, 25.31%. Found: C, 61.74; H, 4.77; N, 25.15%.





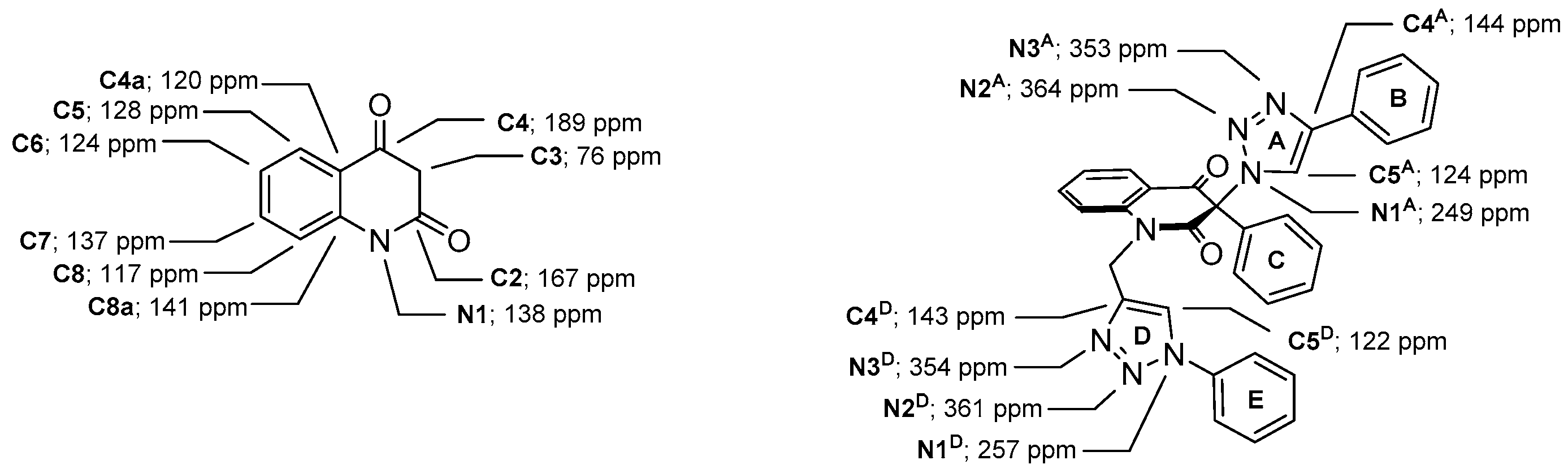

{kind=link}
{kind=link}
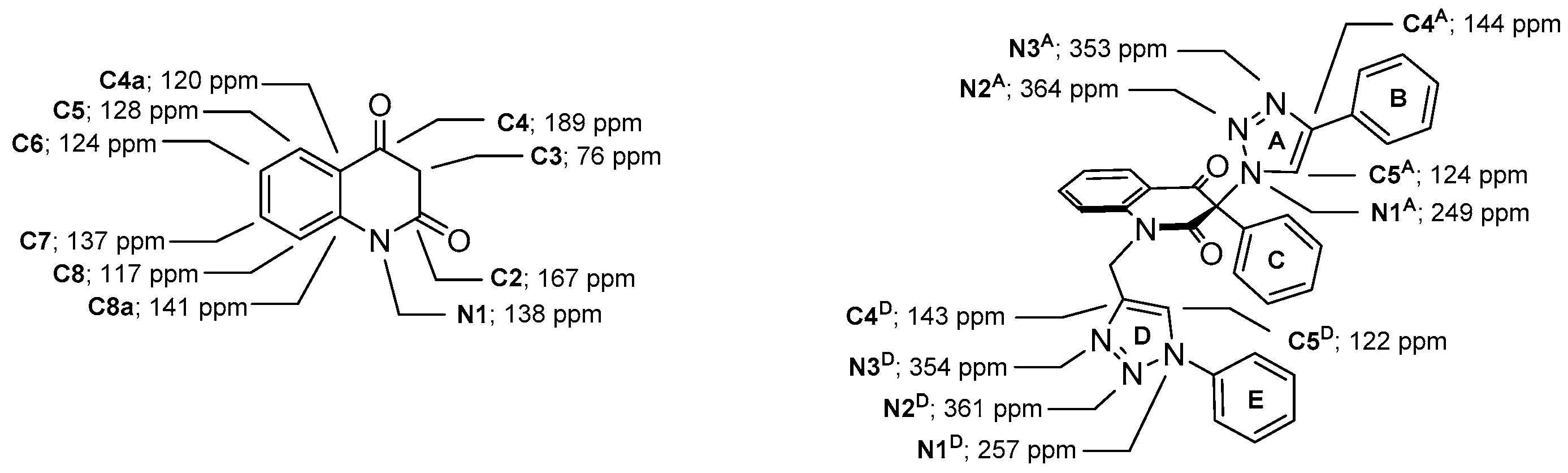{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}