Identfication of Potent LXRβ-Selective Agonists without LXRα Activation by In Silico Approaches

Abstract
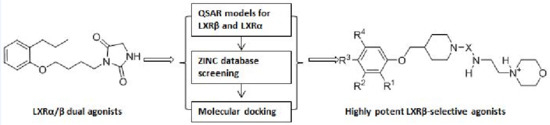
:
1. Introduction
2. Results and Discussion
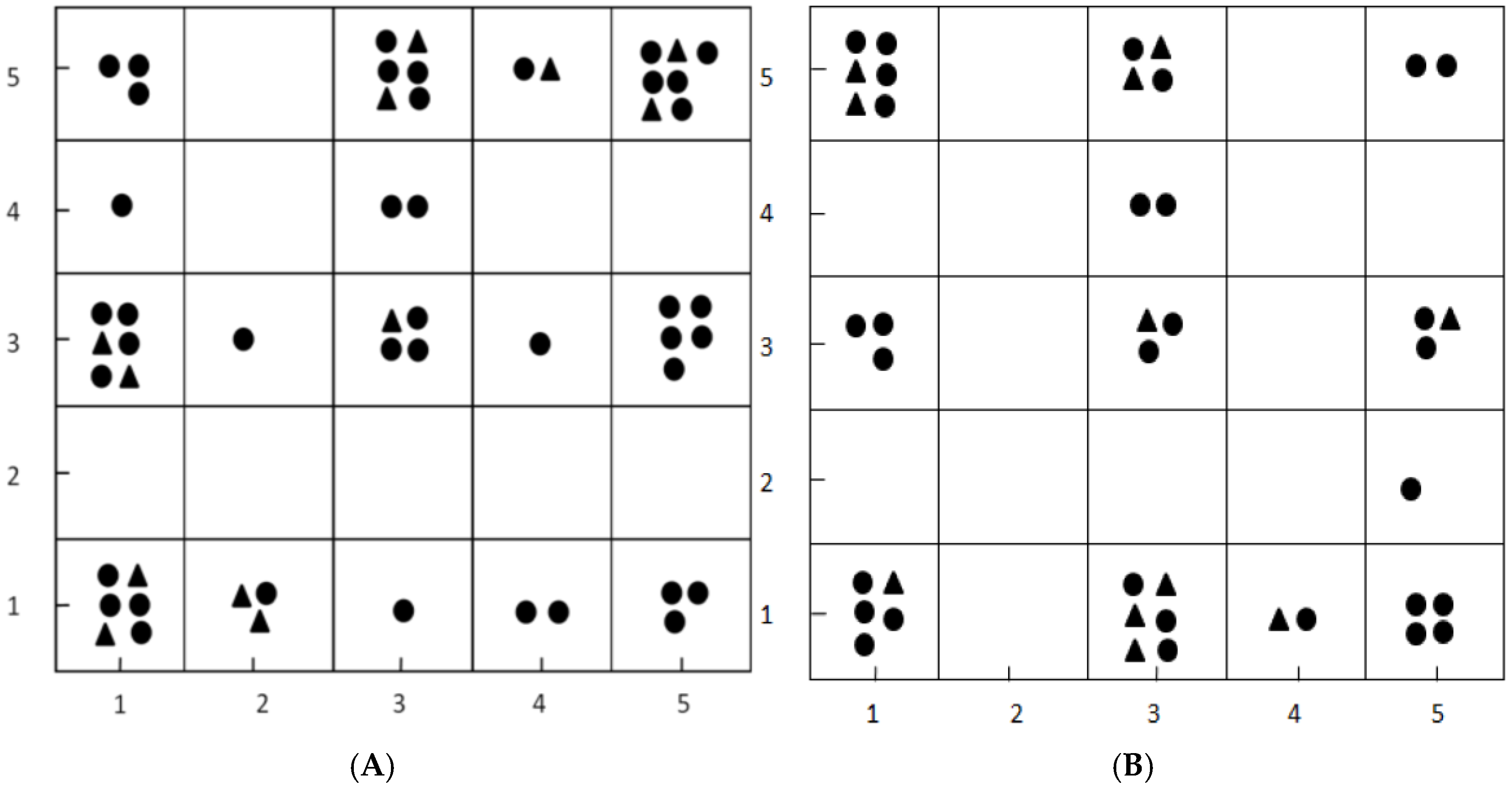
2.1. Results of Dataset Division by Kohonen Map
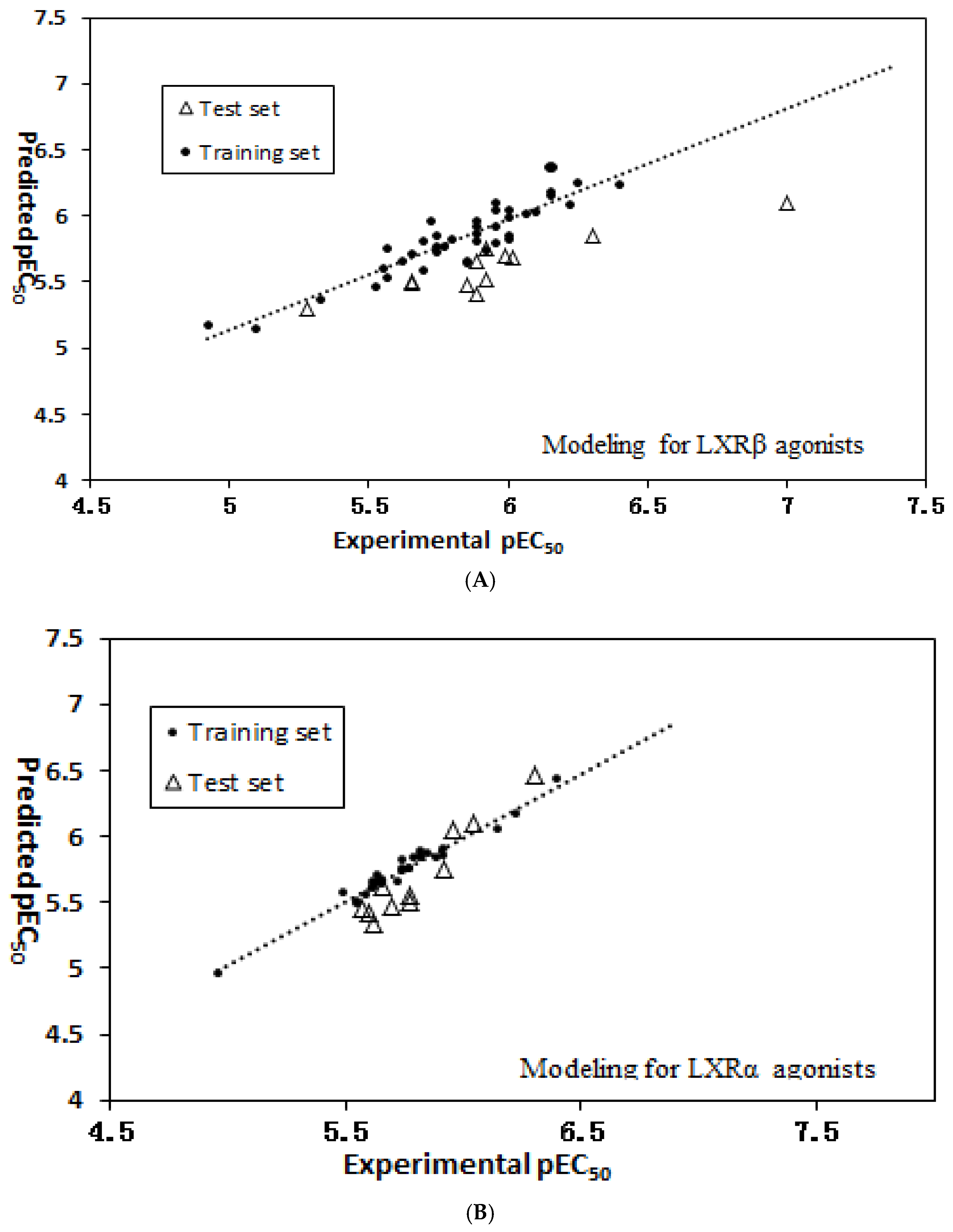
2.2. MLR Model Results of LXRβ Activity
2.3. QSAR Model Results of LXRα Activity
2.4. Interpretation of the Descriptors
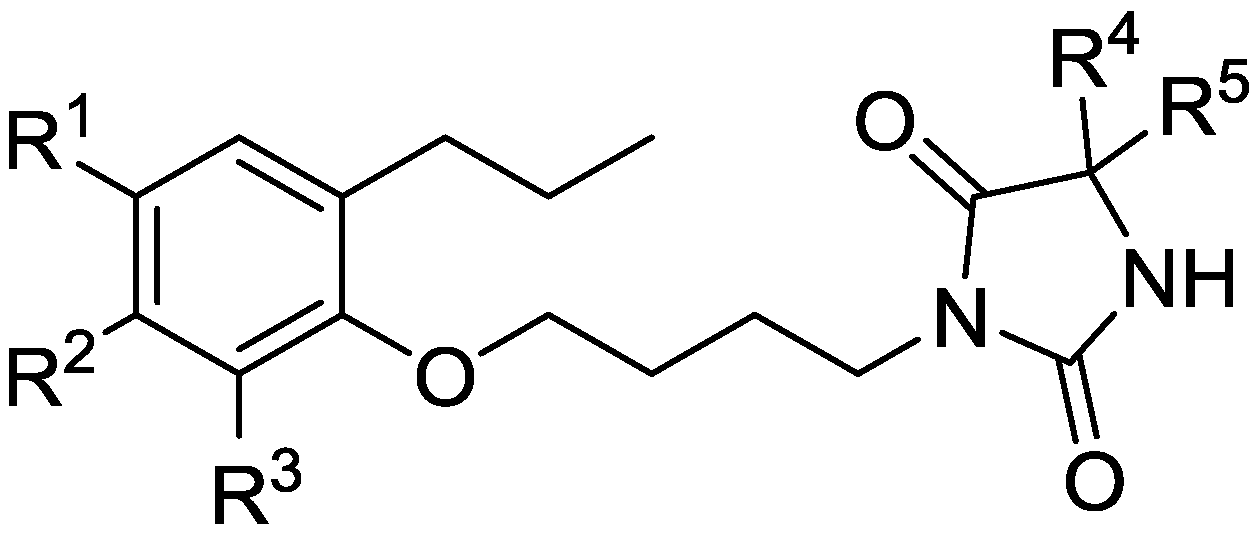
2.5. Screening New Highly LXRβ-Selective Agonists
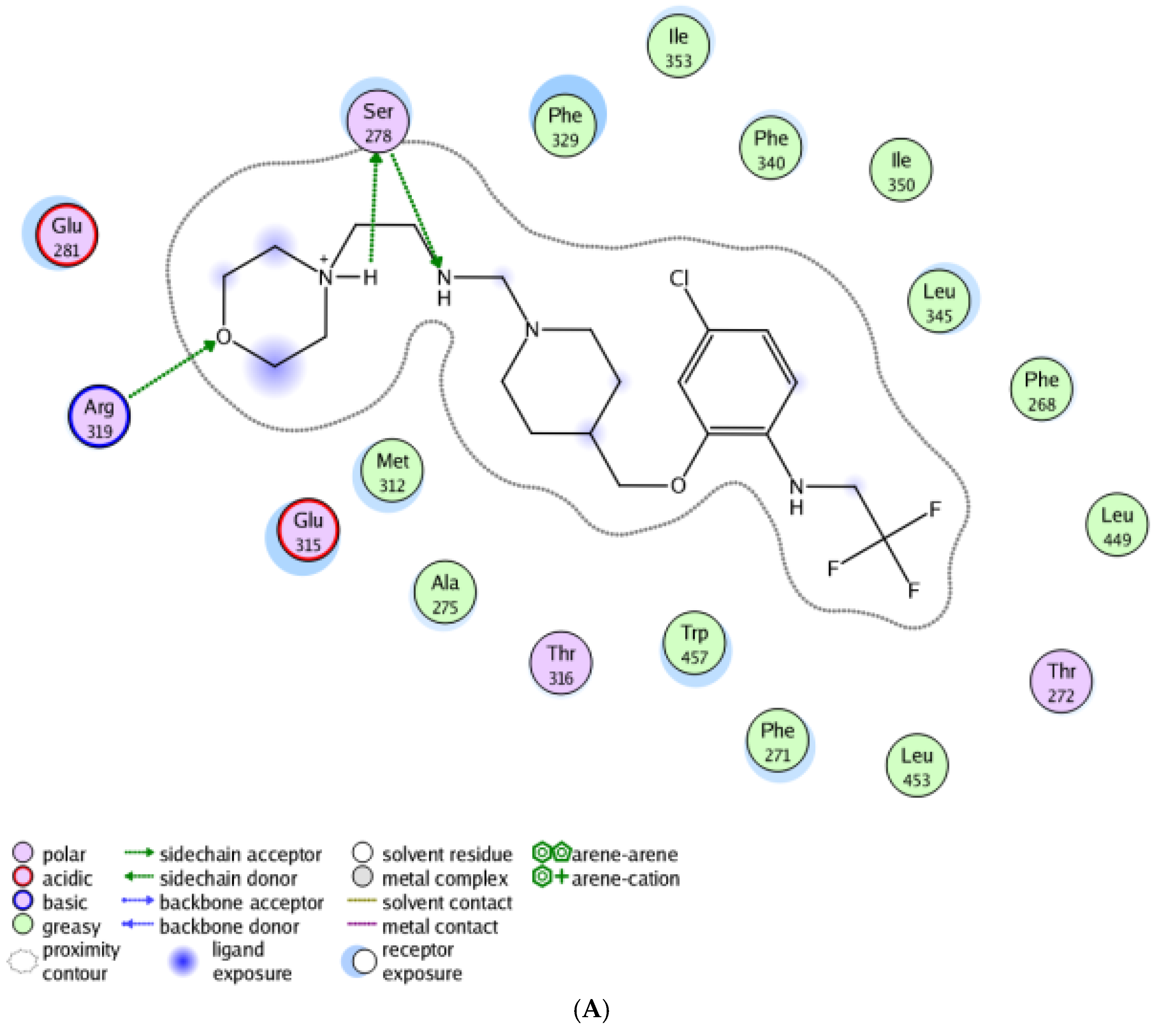
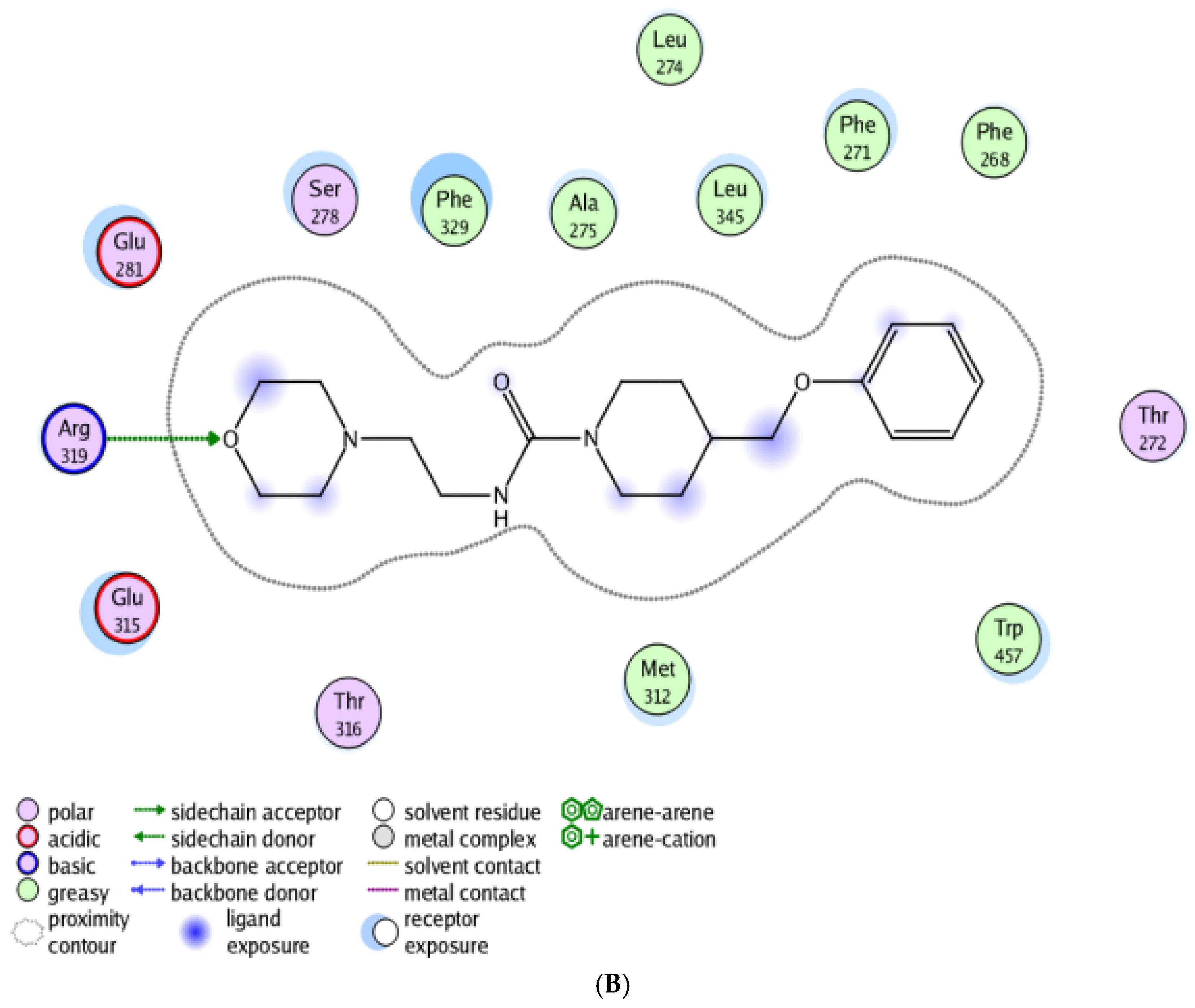
2.6. Molecular Docking Study
3. Materials and Methods
3.1. Dataset Division
3.2. Stepwise Multiple Linear Regression (SW-MLR)
3.3. Model Validation
3.4. Screening News LXRβ-Selective Agonists
3.5. Molecular Docking Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Moriyama, Y.; Okamura, T.; Inazu, A.; Doi, M.; Iso, H.; Mouri, Y.; Ishikawa, Y.; Suzuki, H.; Iida, M.; Koizumi, J.; et al. A low prevalence of coronary heart disease among subjects with increased high-density lipoprotein cholesterol levels, including those with plasma cholesteryl ester transfer protein deficiency. Prev. Med. 2016, 27, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Deng, C.; Hu, W.; Zhou, J.; Fan, C.; Di, S.; Liu, D.; Yang, Y.; Wang, D. Liver X receptors and their agonists: Targeting for cholesterol homeostasis and cardiovascular diseases. Curr. Issues Mol. Biol. 2017, 22, 41–64. [Google Scholar] [CrossRef] [PubMed]
- Breevoort, S.R.; Angdisen, J.; Schulman, I.G. Macrophage-independent regulation of reverse cholesterol transport by liver x receptors. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.K.; Connolly, T.M. Nuclear receptors as drug targets in obesity, dyslipidemia and atherosclerosis. Curr. Opin. Investig. Drugs 2008, 9, 247–255. [Google Scholar] [PubMed]
- Fitz, N.F.; Cronican, A.; Pham, T.; Fogg, A.; Fauq, A.H.; Chapman, R.; Lefterov, I.; Koldamova, R. Liver X receptor agonist treatment ameliorates amyloid pathology and memory deficits caused by high-fat diet in APP23 mice. J. Neurosci. 2010, 30, 6862–6872. [Google Scholar] [CrossRef] [PubMed]
- Gabbi, C.; Warner, M.; Gustafsson, J.Å. Action mechanisms of liver x receptors. Biochem. Biophys. Res. Commun. 2014, 446, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.H.; Yang, J.H.; Shin, B.Y.; Seo, K.; Shin, S.M.; Cho, I.J.; Ki, S.H. Resveratrol inhibits lxrα-dependent hepatic lipogenesis through novel antioxidant sestrin2 gene induction. Toxicol. Appl. Pharm. 2013, 271, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yang, Y.; Yu, Y.; Wen, G.; Shang, N.; Zhuang, W.; Lu, D.; Zhou, B.; Liang, B.; Yue, X.; et al. Synthesis and identification of new flavonoids targeting liver x receptor β involved pathway as potential facilitators of aβ clearance with reduced lipid accumulation. J. Med. Chem. 2013, 56, 6033–6053. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.G.; Peterson, L.B.; Adams, A.D.; Lam, M.N.; Burton, C.A.; Chin, J.; Guo, Q.; Huang, S.; Latham, M.; Lopez, J.C.; et al. Different roles of liver X receptor alpha and beta in lipid metabolism: Effects of an alpha-selective and a dual agonist in mice deficient in each subtype. Biochem. Pharmacol. 2006, 71, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Von Grafenstein, S.; Mihaly-Bison, J.; Wolber, G.; Bochkov, V.N.; Liedl, K.R.; Schuster, D. Identification of Novel Liver X Receptor Activators by Structure-Based Modeling. J. Chem. Inf. Model. 2012, 52, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, L.; Liu, Z.; Li, C.; Xu, J.; Gu, Q.; Xu, J. Predicting selective liver X receptor β agonists using multiple machine learning methods. Mol. Biosyst. 2015, 11, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Liu, Z.; Yan, X.; Ren, J.; Xu, J. A de novo substructure generation algorithm for identifying the privileged chemical fragments of liver x receptorβ agonists. Sci. Rep. 2017, 7, 11121. [Google Scholar] [CrossRef] [PubMed]
- Temml, V.; Voss, C.V.; Dirsch, V.M.; Schuster, D. Discovery of new liver x receptor agonists by pharmacophore modeling and shape-based virtual screening. J. Chem. Inf. Model. 2014, 54, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Hegymegibarakonyi, B.; Orfi, L.; Kéri, G.; Kövesdi, I. Application of Kohonen Self-Organizing feature maps in QSAR of human ADMET and kinase data sets. Acta Pharm. Hung. 2013, 83, 143–148. [Google Scholar]
- Wang, Y.; Chen, J.; Yang, X.; Lyakurwa, F.; Li, X.; Qiao, X. In silico model for predicting soil organic carbon normalized sorption coefficient (K(OC)) of organic chemicals. Chemosphere 2015, 119, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Jalali-Heravi, M.; Asadollahi-Baboli, M.; Shahbazikhah, P. QSAR study of heparanase inhibitors activity using artificial neural networks and Levenberg-Marquardt algorithm. Eur. J. Med. Chem. 2008, 43, 548–556. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Yang, Z.; Ji, G. Feature selection for high-dimensional multi-category data using pls-based local recursive feature elimination. Expert Syst. Appl. 2014, 41, 1463–1475. [Google Scholar] [CrossRef]
- Rastija, V.; Agiä, D.; Tomiå, S.; Nikoliä, S.; Hranjec, M.; Grace, K.Z.; Abramić, M. Synthesis, QSAR, and molecular dynamics simulation of amidino-substituted benzimidazoles as dipeptidyl peptidase iii inhibitors. Acta Chim. Slov. 2015, 62, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Singh, K.P.; Gupta, S.; Rai, P. Predicting acute aquatic toxicity of structurally diverse chemicals in fish using artificial intelligence approaches. Ecotoxicol. Environ. Saf. 2013, 95, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Li, J.; Chen, Z.; Wang, F.; Huang, W.; Hong, Z.; Lin, J. Multistage virtual screening and identification of novel HIV-1 protease inhibitors by integrating SVM, shape, pharmacophore and docking methods. Eur. J. Med. Chem. 2015, 101, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Yang, F.; Yang, X.; Lai, X.; Gao, Y. Systematic understanding of mechanisms of a chinese herbal formula in treatment of metabolic syndrome by an integrated pharmacology approach. Int. J. Mol. Sci. 2016, 17, 2114. [Google Scholar] [CrossRef] [PubMed]
- Koura, M.; Matsuda, T.; Okuda, A.; Watanable, Y.; Yamaguchi, Y.; Kurobuchi, S.; Matsumoto, Y.; Shibuya, K. Design, synthesis and pharmacology of 1,1-bistrifluoromethylcarbinol derivatives as liver X receptor β-selective agonists. Bioorg. Med. Chem. Lett. 2015, 25, 2668–2674. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Okuda, A.; Watanabe, Y.; Miura, T.; Ozawa, H.; Tosaka, A.; Yamazaki, K.; Yamaguchi, Y.; Kurobuchi, S.; Koura, M.; et al. Design and discovery of 2-oxochromene derivatives as liver X receptor β-selective agonists. Bioorg. Med. Chem. Lett. 2015, 25, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Yang, X.; Lai, X.; Gao, Y. 2D and 3D QSAR models for identifying diphenylpyridylethanamine based inhibitors against cholesteryl ester transfer protein. Bioorg. Med. Chem. Lett. 2015, 25, 4487–4495. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Yang, X.; Lai, X.; Kang, J.; Gan, H.; Gao, Y. Structural Investigation for Optimization of Anthranilic Acid Derivatives as Partial FXR Agonists by in Silico Approaches. Int. J. Mol. Sci. 2016, 17, 536. [Google Scholar] [CrossRef] [PubMed]
- Toropov, A.A.; Toropova, A.P. Quasi-QSAR for mutagenic potential of multi-walled carbon-nanotubes. Chemosphere 2015, 124, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Ghorbanzadeh, M.; Zhang, J.; Andersson, P.L. Binary classification model to predict developmental toxicity of industrial chemicals in zebrafish. J. Chemometr. 2016, 30, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Bolboacă, S.D.; Jäntschi, L. Sensitivity, specificity, and accuracy of predictive models on phenols toxicity. J. Comput. Sci. 2014, 5, 345–350. [Google Scholar] [CrossRef]
- Zou, S.; Zhang, J.; Zhang, Z. A novel approach for predicting microbe-disease associations by bi-random walk on the heterogeneous network. PLoS ONE 2017, 12, e0184394. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegazy, M.F.; Ibrahim, A.Y.; Mohamed, T.A.; Shahat, A.A.; El Halawany, A.M.; Abdelazim, N.; Alsaid, M.S.; Paré, P.W. Sesquiterpene lactones from cynara cornigera: Acetyl cholinesterase inhibition and in silico ligand docking. Planta Med. 2016, 82, 138–146. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Descriptor | Chemical Meaning | Coefficient | Standard Coefficient | VIF | p-Value |
|---|---|---|---|---|---|
| vsurf_IW2 | Hydrophilic integy moment | −0.777 | −0.592 | 2.246 | 0.000 |
| SMR_VSA6 | Sum of vi such that Ri is in (0.485,0.56] | 0.007 | 0.325 | 1.232 | 0.000 |
| glob | Globularity, or inverse condition number (smallest eigenvalue divided by the largest eigenvalue) of the covariance matrix of atomic coordinates. | −1.236 | −0.668 | 1.779 | 0.000 |
| GCUT_SLOGP_2 | The GCUT descriptors using atomic contribution to logP | 4.560 | 0.687 | 2.785 | 0.000 |
| E_strain | Local strain energy | −60.185 | −0.315 | 1.196 | 0.000 |
| dipoleX | The x component of the dipole moment | −0.189 | −0.358 | 1.240 | 0.000 |
| AM1_LUMO | The energy (eV) of the Lowest Unoccupied Molecular Orbital calculated using the AM1 Hamiltonian | −0.247 | −0.397 | 2.712 | 0.003 |
| vsurf_IW5 | Hydrophilic integy moment | 0.154 | 0.284 | 1.790 | 0.007 |
| vsurf_DD13 | Contact distances of vsurf_DDmin | 0.016 | 0.212 | 1.192 | 0.013 |
| Constant | 5.087 |
| Training Set | Test Set | ||||||
|---|---|---|---|---|---|---|---|
| QSARModel | R2train | RMSEtrain | F | Q2LOO | RMSELOO | R2test | RMSEtest |
| LXR beta | 0.837 | 0.118 | 17.235 | 0.715 | 0.156 | 0.843 | 0.232 |
| LXR alpha | 0.968 | 0.045 | 44.068 | 0.895 | 0.081 | 0.914 | 0.155 |
| Descriptor | Chemical Meaning | Coefficient | Standard Coefficient | VIF | p-Value |
|---|---|---|---|---|---|
| GCUT_SLOGP_2 | The GCUT descriptors using atomic contribution to logP | 8.952 | 1.539 | 4.862 | 0.000 |
| vsurf_DD12 | Contact distances of vsurf_DDmin | 0.024 | 0.366 | 1.348 | 0.000 |
| Q_VSA_POS | Total positive van der Waals surface area | 0.005 | 0.673 | 2.161 | 0.000 |
| SlogP_VSA2 | Sum of vi such that Li is in (−0.2,0] | 0.023 | 0.813 | 4.763 | 0.000 |
| E_ang | Angle bend potential energy | −0.019 | −0.325 | 2.126 | 0.000 |
| pmiY | y component of the principal moment of inertia | 4.808 × 10−5 | 0.209 | 1.468 | 0.001 |
| dipoleY | The y component of the dipole moment | 0.164 | 0.281 | 1.408 | 0.000 |
| vsurf_DW12 | Contact distances of vsurf_EWmin | −0.019 | −0.203 | 1.467 | 0.001 |
| BCUT_SMR_0 | The BCUT descriptors using atomic contribution to molar refractivity | 34.772 | 0.351 | 2.546 | 0.000 |
| SlogP_VSA3 | Sum of vi such that Li is in (0,0.1] | 0.005 | 0.238 | 1.590 | 0.000 |
| vsurf_CW6 | Capacity factor | −4.271 | −0.273 | 3.370 | 0.003 |
| Q_VSA_FPPOS | Fractional positive polar van der Waals surface area | −1.339 | −0.144 | 1.810 | 0.024 |
| Constant | 83.858 |
| No. of Test | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| LXRβ model | R2train | 0.185 | 0.182 | 0.155 | 0.257 | 0.282 | 0.208 | 0.126 | 0.244 | 0.155 | 0.241 |
| Q2LOO | 0.063 | 0.006 | 0.018 | 0.001 | 0.037 | 0.015 | 0.081 | 0.002 | 0.047 | 0.003 | |
| LXRα model | R2train | 0.259 | 0.174 | 0.288 | 0.287 | 0.334 | 0.298 | 0.25 | 0.264 | 0.258 | 0.287 |
| Q2LOO | 0.033 | 0.097 | 0.086 | 0.009 | 0.015 | 0.039 | 0.023 | 0.016 | 0.032 | 0.027 | |
| AM1_LUMO | GCUT_SLOGP_2 | E_strain | dipoleX | SMR_VSA6 | vsurf_DD13 | vsurf_IW2 | vsurf_IW5 | Glob | |
|---|---|---|---|---|---|---|---|---|---|
| BCUT_SMR_0 | 0.031 | −0.255 | 0.254 | 0.199 | −0.243 | −0.172 | 0.075 | −0.069 | −0.063 |
| GCUT_SLOGP_2 | −0.055 | 0.014 | 0.066 | −0.194 | −0.371 | 0.090 | 0.226 | −0.133 | 0.110 |
| Q_VSA_FPPOS | −0.242 | −0.315 | 0.072 | −0.008 | −0.041 | 0.258 | −0.178 | −0.184 | −0.117 |
| Q_VSA_POS | −0.017 | 0.279 | −0.182 | −0.164 | 0.240 | 0.113 | −0.202 | −0.012 | 0.013 |
| E_ang | −0.069 | −0.431 | 0.007 | −0.030 | 0.173 | 0.531 | 0.148 | −0.261 | −0.361 |
| dipoleY | −0.216 | 0.045 | −0.168 | −0.189 | 0.051 | −0.001 | −0.062 | −0.086 | 0.254 |
| pmiY | 0.204 | −0.159 | −0.195 | −0.098 | −0.020 | 0.110 | 0.216 | 0.067 | −0.353 |
| SlogP_VSA2 | 0.029 | −0.166 | −0.012 | 0.083 | 0.210 | 0.054 | −0.076 | 0.051 | −0.347 |
| SlogP_VSA3 | −0.155 | 0.246 | 0.095 | −0.032 | −0.236 | 0.015 | 0.026 | 0.082 | 0.085 |
| vsurf_CW6 | −0.056 | 0.002 | 0.352 | −0.231 | −0.116 | −0.127 | −0.137 | −0.156 | 0.201 |
| vsurf_DD12 | −0.564 | 0.005 | 0.161 | 0.067 | −0.400 | 0.226 | 0.243 | 0.060 | −0.211 |
| vsurf_DW12 | 0.116 | −0.075 | −0.046 | −0.324 | 0.099 | −0.099 | −0.092 | −0.304 | −0.091 |
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Name | R1 | R2 | R3 | R4 | X | Predicted pEC50 Values | ||
| LXRβ | LXRα | Docking Scores | ||||||
| ZINC55084484 | H | H | H | H | CO | 7.343 | −1.901 | −7.713 |
| N1 | 2,2,2-trifluoroethylamino | H | H | Cl | CH2 | 8.497 | −1.911 | −11.205 |
| N2 | 2,2,2-trifluoroethylamino | H | H | H | CH2 | 8.390 | −2.076 | −9.394 |
| N3 | 2,2,2-trifluoroethylamino | Cl | H | H | CH2 | 8.429 | −1.730 | −9.757 |
| N4 | 2,2,2-trifluoroethylamino | F | H | H | CO | 8.328 | 0.215 | −10.236 |
| N5 | propionyloxy | H | H | H | CO | 8.148 | −0.753 | −9.528 |
| N6 | 2,2,2-trifluoroethylamino | H | propionyloxy | H | CH2 | 7.932 | −0.9524 | −9.323 |
| N7 | propionyloxy | H | propionyloxy | H | CO | 7.923 | −0.905 | −9.177 |
| N8 | 2,2,2-trifluoroethylamino | F | H | H | CH2 | 8.178 | −1.760 | −10.068 |
| N9 | 2,2,2-trifluoroethylamino | H | propionyloxy | H | CO | 8.111 | −1.211 | −10.321 |
| Name | Predicted pEC50 Values | Weight | a_acc | a_don | logP(o/w) | |
|---|---|---|---|---|---|---|
| LXRβ | LXRα | |||||
| ZINC55084484 | 7.343 | −1.901 | 347.459 | 4 | 1 | 1.218 |
| N1 | 8.497 | −1.911 | 465.968 | 4 | 2 | 2.757 |
| N2 | 8.390 | −2.076 | 431.523 | 4 | 2 | 2.128 |
| N3 | 8.429 | −1.730 | 465.968 | 4 | 2 | 2.718 |
| N4 | 8.328 | 0.215 | 463.496 | 3 | 2 | 1.777 |
| N5 | 8.148 | −0.753 | 420.53 | 4 | 1 | 1.376 |
| N6 | 7.932 | −0.9524 | 503.586 | 5 | 2 | 2.573 |
| N7 | 7.923 | −0.905 | 492.593 | 5 | 1 | 1.821 |
| N8 | 8.178 | −1.760 | 449.513 | 4 | 2 | 2.279 |
| N9 | 8.111 | −1.211 | 517.569 | 4 | 2 | 2.071 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, M.; Yang, F.; Kang, J.; Gan, H.; Yang, X.; Lai, X.; Gao, Y. Identfication of Potent LXRβ-Selective Agonists without LXRα Activation by In Silico Approaches. Molecules 2018, 23, 1349. https://doi.org/10.3390/molecules23061349
Chen M, Yang F, Kang J, Gan H, Yang X, Lai X, Gao Y. Identfication of Potent LXRβ-Selective Agonists without LXRα Activation by In Silico Approaches. Molecules. 2018; 23(6):1349. https://doi.org/10.3390/molecules23061349
Chicago/Turabian StyleChen, Meimei, Fafu Yang, Jie Kang, Huijuan Gan, Xuemei Yang, Xinmei Lai, and Yuxing Gao. 2018. "Identfication of Potent LXRβ-Selective Agonists without LXRα Activation by In Silico Approaches" Molecules 23, no. 6: 1349. https://doi.org/10.3390/molecules23061349
APA StyleChen, M., Yang, F., Kang, J., Gan, H., Yang, X., Lai, X., & Gao, Y. (2018). Identfication of Potent LXRβ-Selective Agonists without LXRα Activation by In Silico Approaches. Molecules, 23(6), 1349. https://doi.org/10.3390/molecules23061349