Three New Highly Oxygenated Germacranolides from Carpesium Divaricatum and Their Cytotoxic Activity


Abstract
:1. Introduction
2. Results and Discussion
2.1. Purification of Compounds 1–5
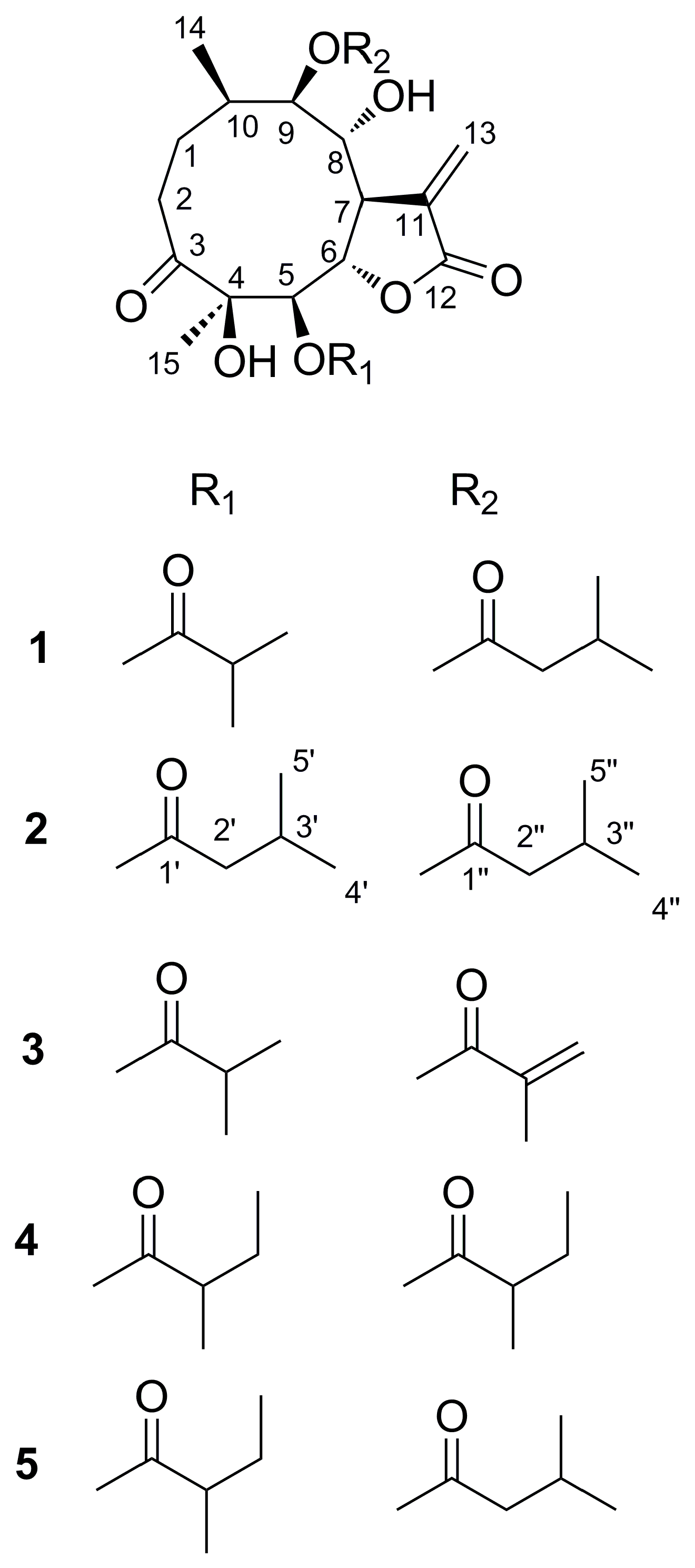
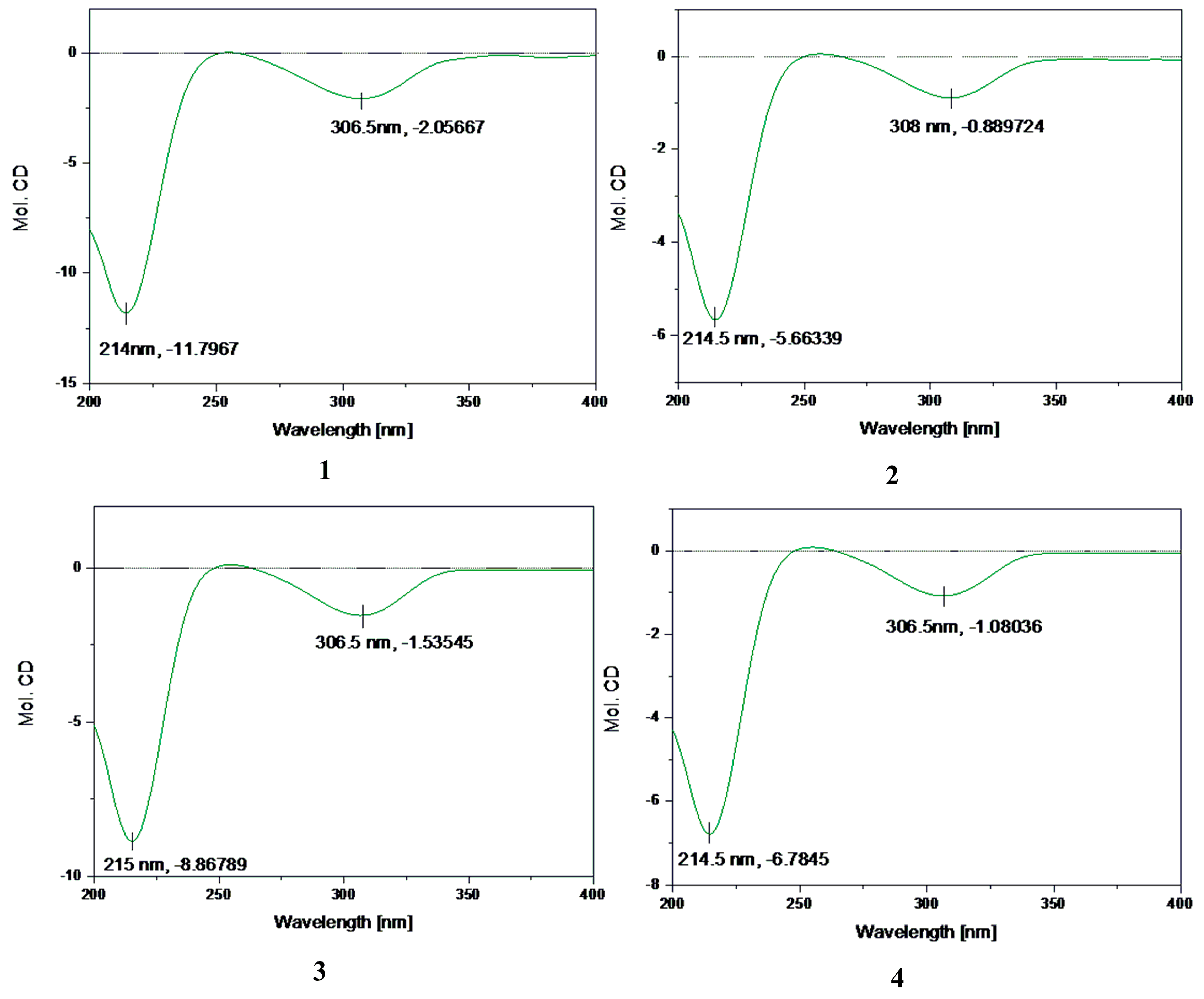
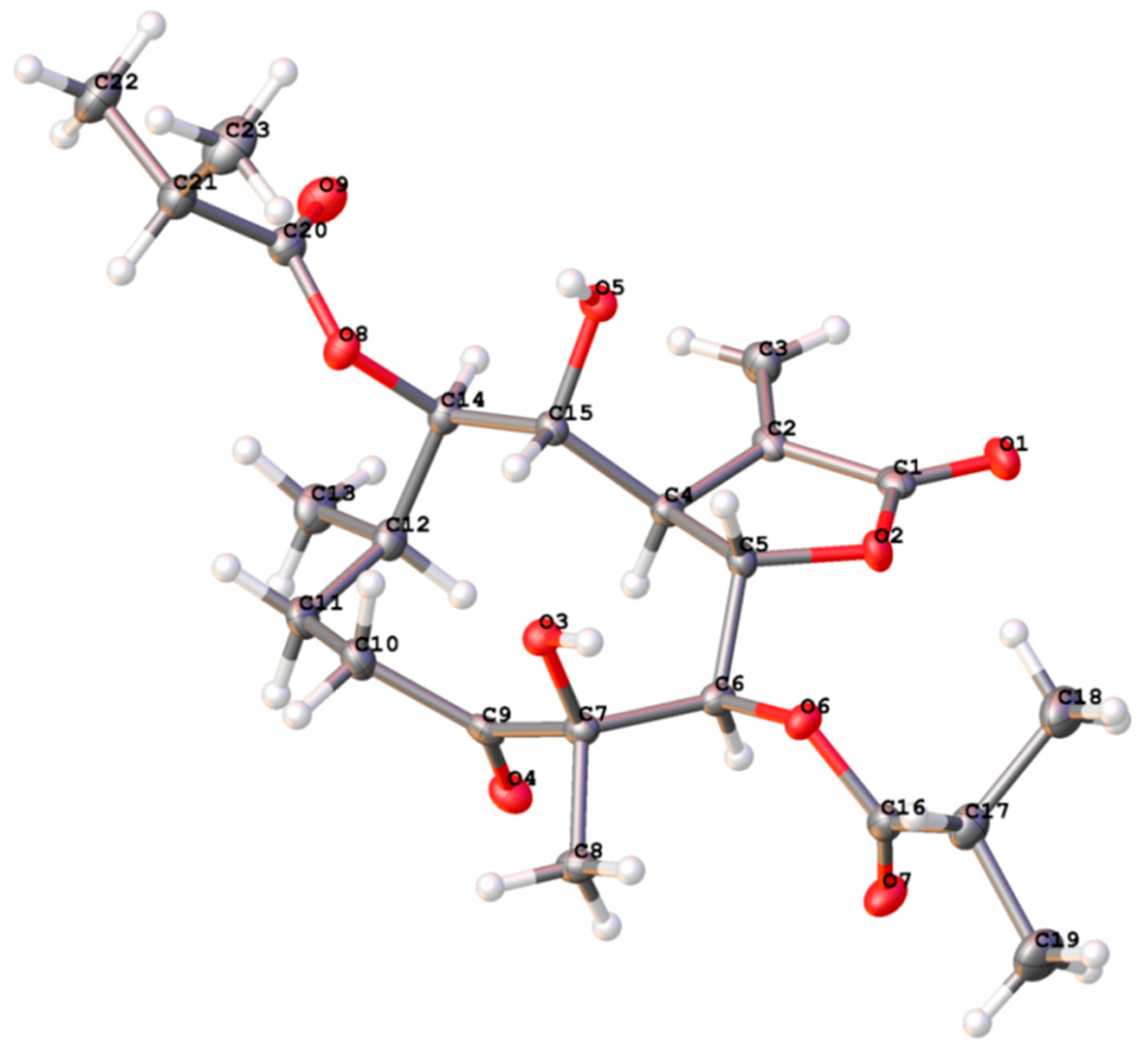
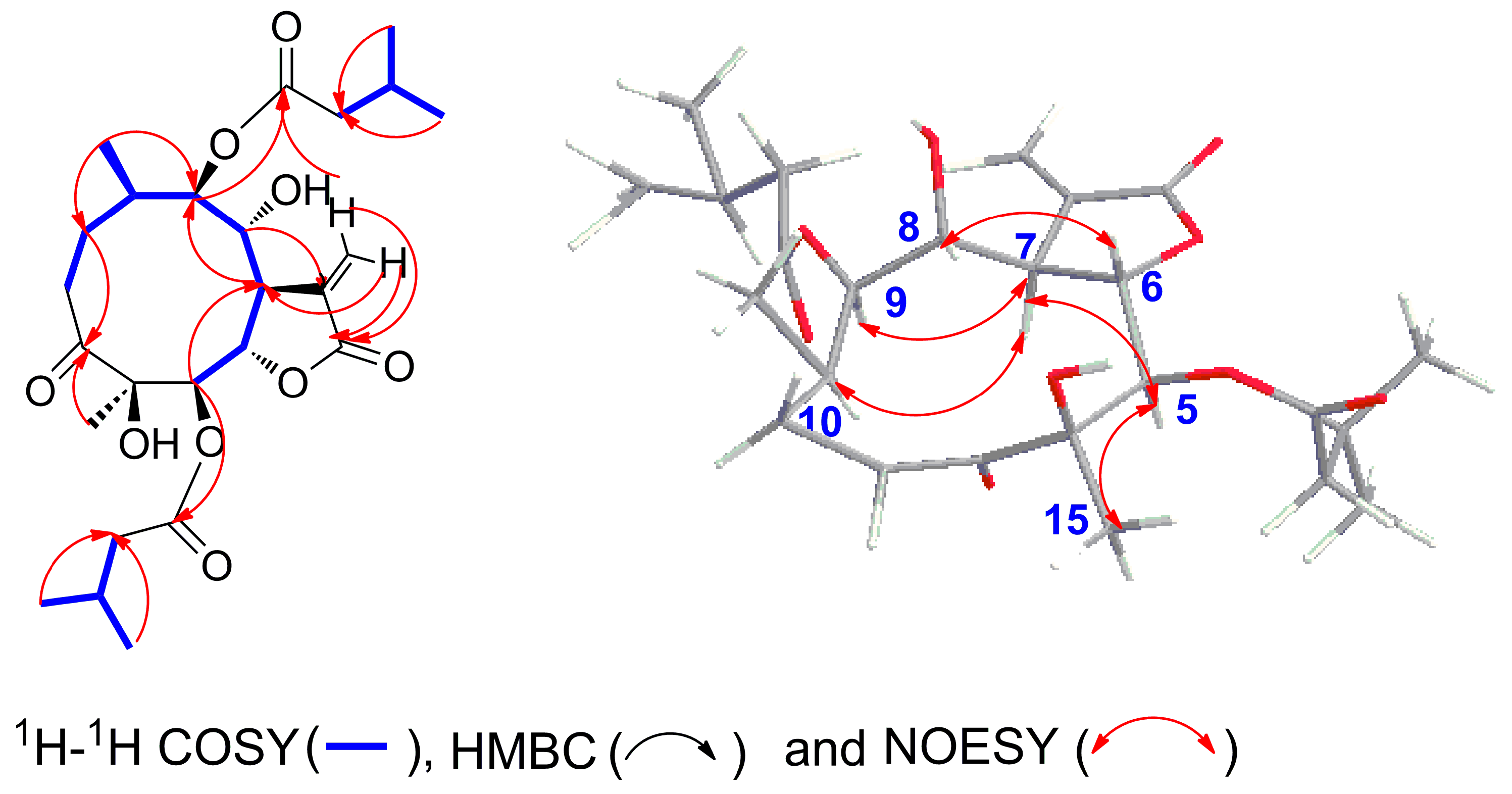
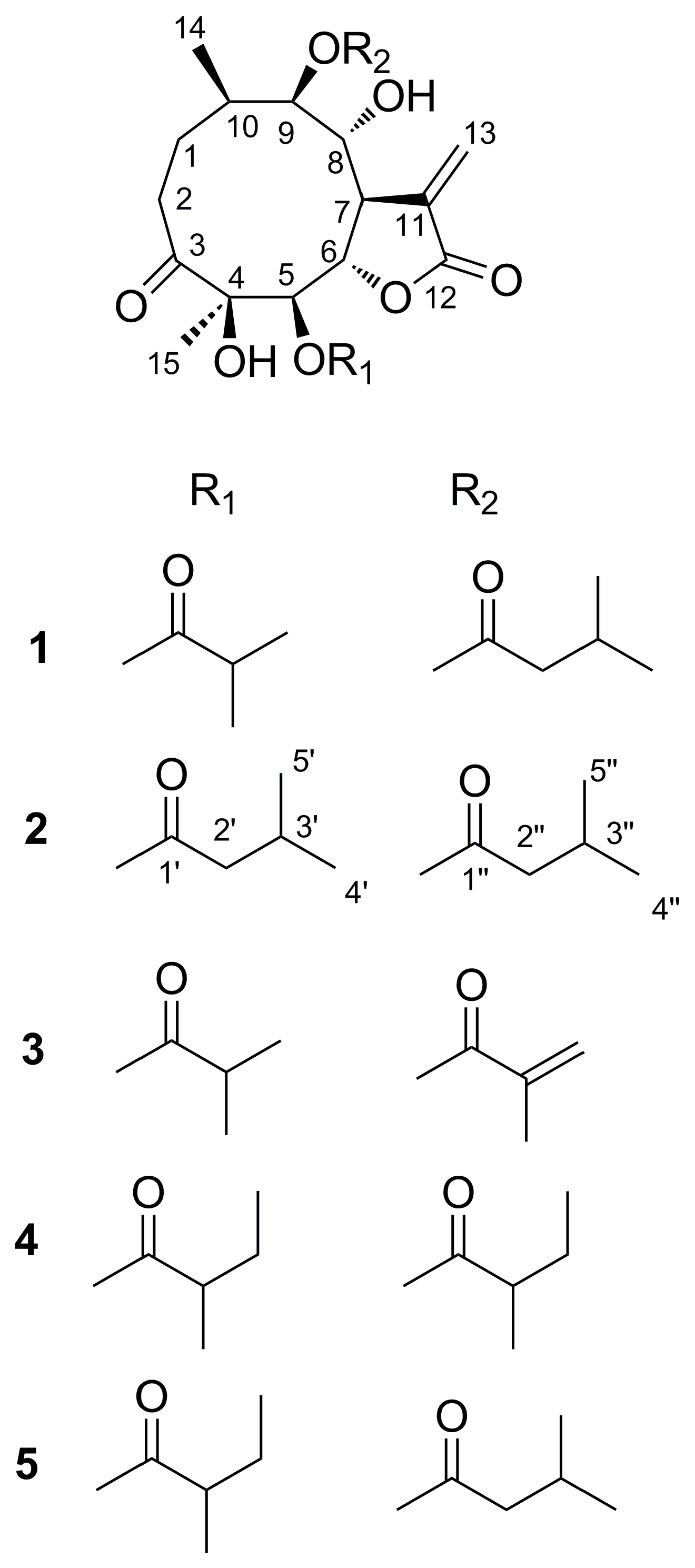
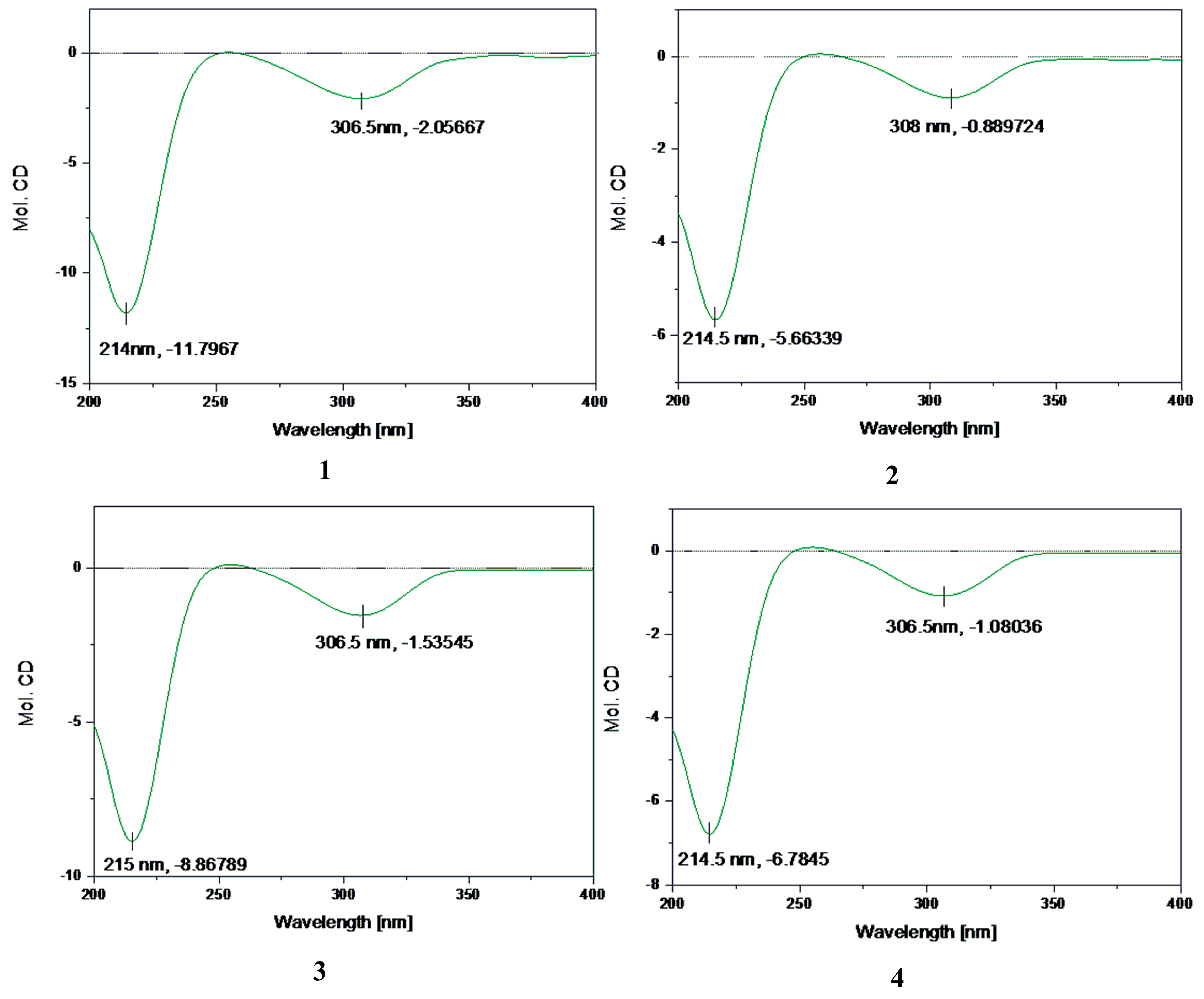
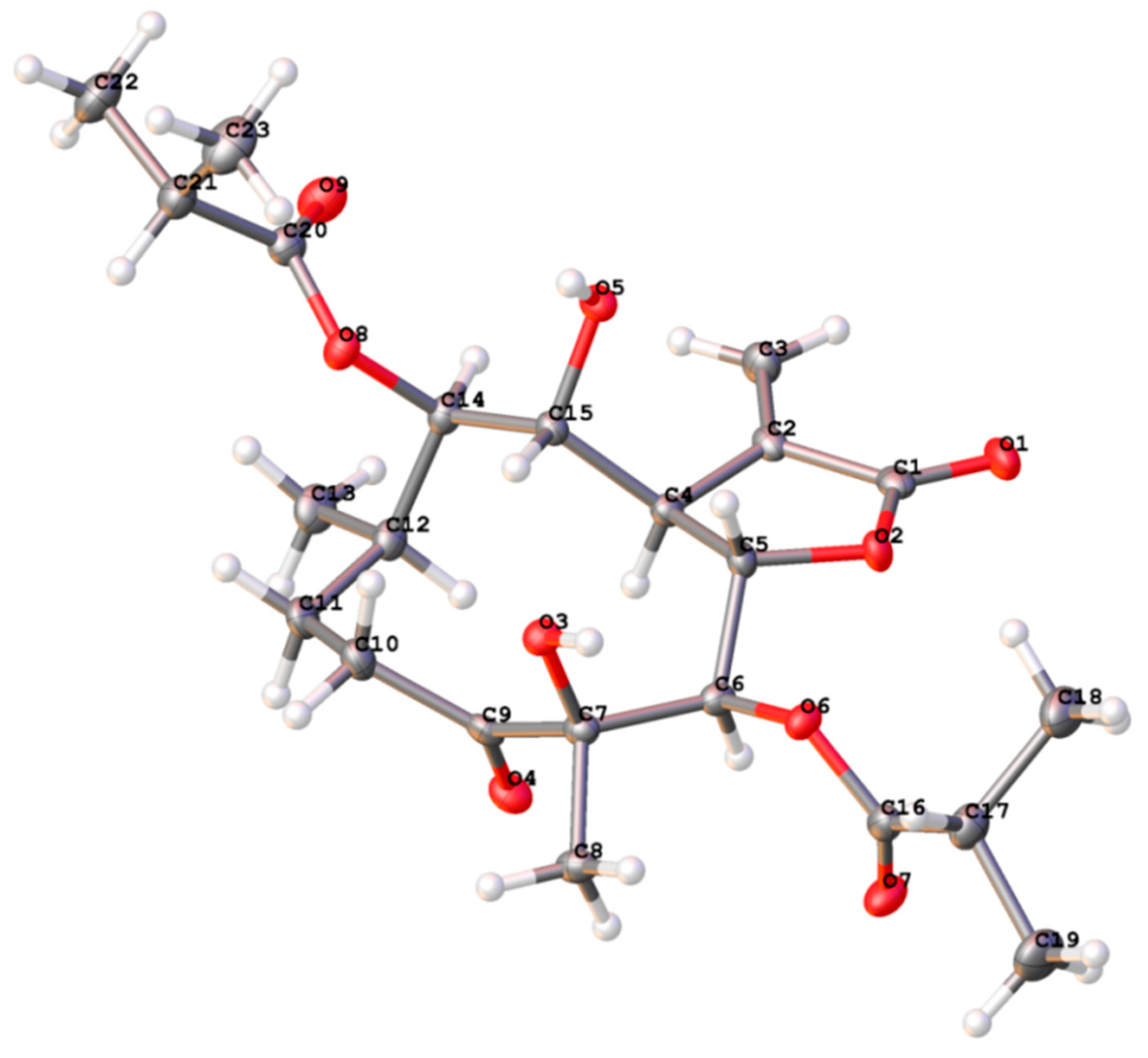
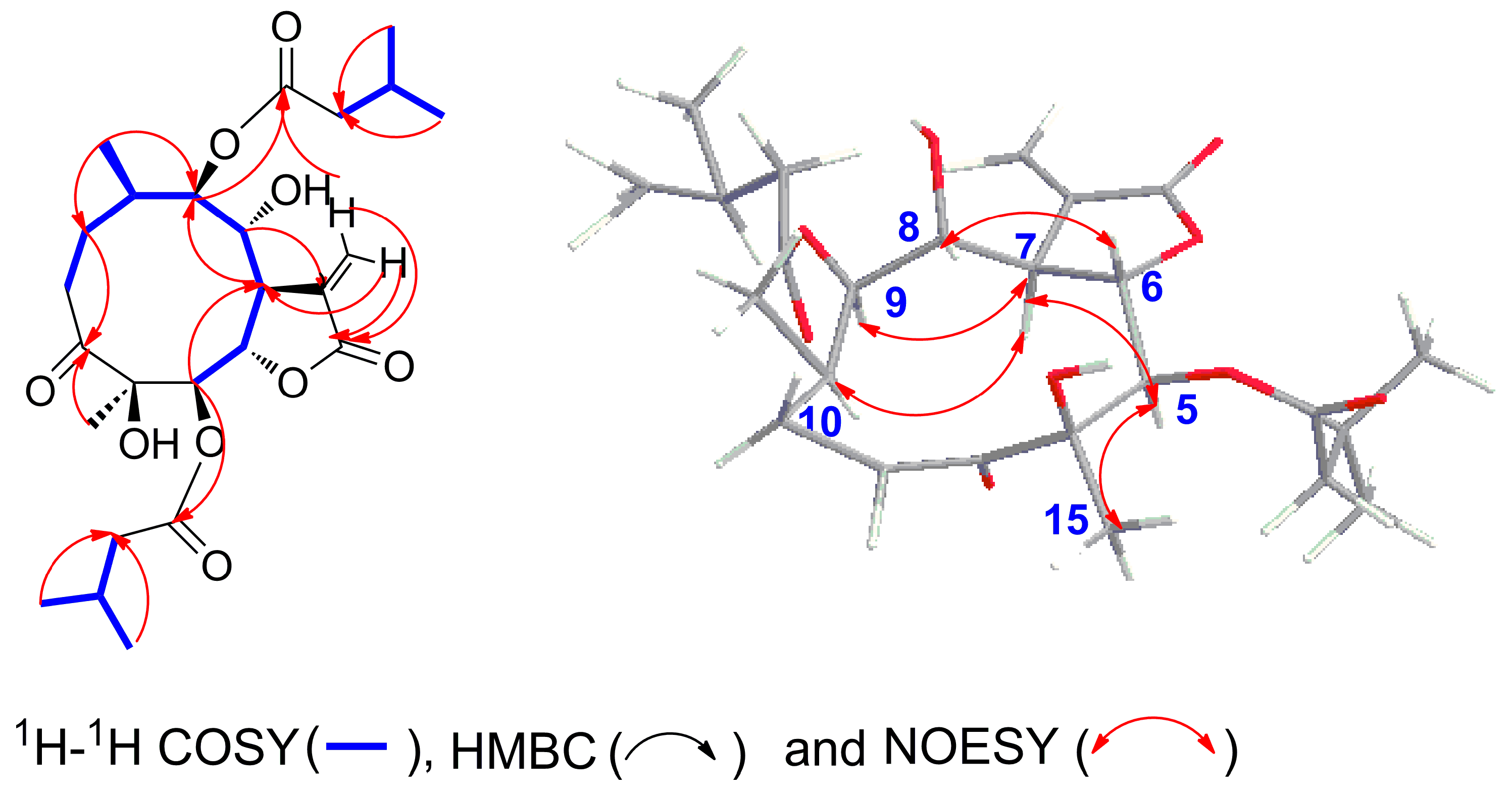
2.2. Structure Elucidation of Compounds 1–5
2.3. In Vitro Cytotoxic Activities of Compounds 1–5
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Isolation and Purification of Compounds 1–5
3.4. Characterization of Compounds 2–4
3.5. X-ray Crystal Structure Analysis of Compound 1
3.6. Cytotoxicity Assays of Compounds 1–5
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Mabberley, D.J. Mabberley′s Plant Book, 3rd ed.; Cambridge University Press: Cambridge, UK, 2008; p. 154. [Google Scholar]
- Zhang, J.P.; Wang, G.W.; Tian, X.H.; Yang, Y.X.; Liu, Q.X.; Chen, L.P.; Li, H.L.; Zhang, W.D. The genus Carpesium: A review of its ethnopharmacology, phytochemistry and pharmacology. J. Ethnopharmacol. 2015, 163, 173–191. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Yuan, C.S.; Jia, Z.J. Xanthanolides, germacranolides, and other constituents from Carpesium longifolium. J. Nat. Prod. 2003, 66, 1554–1557. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.D.; Wang, X.R.; Ma, L.S.; Li, X.; Row, K.H. Sesquiterpenoids from Carpesium divaricatum and their cytotoxic activity. Fitoterapia 2012, 83, 1351–1355. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lin, C.J.; Jia, Z.J. Cytotoxic germacranolides and acyclic diterpenoides from the seeds of Carpesium triste. J. Nat. Prod. 2007, 70, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Ou, J.C. Napalolides A–D, four new sesquiterpene lactones from Carpesium nepalense. J. Nat. Prod. 1996, 59, 991–993. [Google Scholar] [CrossRef]
- Li, X.W.; Weng, L.; Gao, X.; Zhao, Y.; Pang, F.; Liu, J.H.; Zhang, H.F.; Hu, J.F. Antiproliferative and apoptotic sesquiterpene lactones from Carpesium faberi. Bioorg. Med. Chem. Lett. 2011, 21, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.X.; Shan, L.; Liu, Q.X.; Shen, Y.H.; Zhang, J.P.; Ye, J.; Xu, X.K.; Li, H.J.; Zhang, W.D. Carpedilactones A-D, four new isomeric sesquiterpene lactone dimers with potent cytotoxicity from Carpesium faberi. Org. Lett. 2014, 16, 4216–4219. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Tang, C.P.; Chen, L.; Qiao, Y.; Geng, M.Y.; Ye, Y. Dicarabrones A and B, a pair of new epimers dimerized from sesquiterpene lactones via a [3 + 2] cycloaddition from Carpesium abrotanoides. Org. Lett. 2015, 17, 1656–1659. [Google Scholar] [CrossRef] [PubMed]
- Editorial Committee of the Administration Bureau of Traditional Chinese Medicine. Chinese Materia Medica; Shanghai Science &Technology Press: Shanghai, China, 1999; Volume 21, p. 761. [Google Scholar]
- Kim, E.J.; Jin, H.K.; Kim, Y.K.; Lee, H.Y.; Lee, S.Y.; Lee, K.R.; Zee, O.P.; Han, J.W.; Lee, H.W. Suppression by a sesquiterpene lactone from Carpesium divaricatum of inducible nitric oxide synthase by inhibiting nuclear factor-κB activation. Biochem. Pharmacol. 2001, 61, 903–910. [Google Scholar] [CrossRef]
- Zee, O.P.; Kim, D.K.; Choi, S.U.; Lee, C.O.; Lee, K.R. A new cytotoxic acyclic diterpene from Carpesium divaricatum. Arch. Pharm. Res. 1999, 22, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Zee, O.P.; Kim, D.K.; Lee, K.R. Thymol derivatives from Carpesium divaricatum. Arch. Pharm. Res. 1998, 21, 618–620. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.M.; Seo, S.H.; Kang, E.Y.; Park, W.H.; Park, S.D.; Moon, H.I. Antiplasmodial activity of isolated compounds from Carpesium divaricatum. Phytother. Res. 2010, 24, 451–453. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Beak, N.I.; Choi, S.U.; Lee, C.O.; Lee, K.R.; Zee, O.P. Four new cytotoxic germacranolides from Carpesium divaricatum. J. Nat. Prod. 1997, 60, 1199–1202. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, M. Sesquiterpene lactones from Carpesium divaricatum. Phytochemistry 1990, 29, 547–550. [Google Scholar] [CrossRef]
- Kim, D.K.; Lee, K.R.; Zee, O.P. Sesquiterpene lactones from Carpesium divaricatum. Phytochemistry 1997, 46, 1245–1247. [Google Scholar] [CrossRef]
- Lee, H.J.; Jung, H.; Kwon, J.; Li, H.; Lee, D.Y.; Lim, H.J.; Kim, M.R.; Moon, D.C.; Ryu, J.H. A germacranolide sesquiterpene lactone suppressed inducible nitric oxide synthase by downregulating NF-κB activity. Can. J. Physiol. Pharmacol. 2011, 89, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.I.; Zee, O. Antiproliferative effect from sesquiterpene lactones of Carpesium rosulatum MlQ consumed in South Korea on the five human cancer cell lines. Rec. Nat. Prod. 2010, 4, 3149–3155. [Google Scholar]
- Moon, H.I.; Zee, O. Sesquiterpene lactones from Carpesium rosulatum with potential cytotoxicity against five human cancer cell lines. Hum. Exp. Toxicol. 2010, 30, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Baruah, R.N.; Sharma, R.P. Thyagarajan, G. Unusual germacranolides from Inula eupatorioides. J. Org. Chem. 1980, 45, 4838–4843. [Google Scholar] [CrossRef]
- Baruah, N.C.; Baruah, R.N.; Sharma, R.P.; Baruah, J.N. Germacranolides of Inula eupatorioides. 2. Absolute configuration of the ineupatorolides. J. Org. Chem. 1982, 47, 137–140. [Google Scholar] [CrossRef]
- Goswami, A.C.; Baruah, R.N.; Sharma, R.P.; Baruah, J.N.; Kulanthaivel, P.; Herz, W. Germacranolides from Inula cappa. Phytochemistry 1984, 23, 367–372. [Google Scholar] [CrossRef]
- Wang, F.Y.; Li, X.Q.; Sun, Q.; Yao, S.; Ke, C.Q.; Tang, C.P.; Liu, H.C.; Geng, M.Y.; Ye, Y. Sesquiterpene lactones from Carpesium divaricatum. Phytochem. Lett. 2012, 5, 639–642. [Google Scholar] [CrossRef]
- Gonzalez, A.G.; Barrera, J.B.; Mendez, J.T.; Martinez, J.E.; Sanchez, M.L. Germacranolides from Allagopappus viscosissimus. Phytochemistry 1992, 31, 330–331. [Google Scholar] [CrossRef]
- Gonzalez, A.G.; Bermejo, J. Sesquiterpene lactones and other constituents of Allagopappus species. J. Nat. Prod. 1995, 58, 432–437. [Google Scholar] [CrossRef]
- Zhang, T.; Si, J.G.; Zhang, Q.B.; Ding, G.; Zou, Z.M. New highly oxygenated germacranolides from Carpesium divaricatum and their cytotoxic activity. Sci. Rep. 2016, 6, 27237. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.J.; Won, S.J.; Su, Z.R.; Yu, C.L. Free radical scavenging and apoptotic effects of cordyceps sinensis fractionated by supercritical carbon dioxide. Food Chem. Toxicol. 2005, 43, 543–552. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1 and 5 are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 2 a | 3 b | 4 a | |||
|---|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δc, Type | δH (J in Hz) | δc, Type | δH (J in Hz) | |
| 1 | 25.3 CH2 | 1.79 m, 1.65 m | 25.3 CH2 | 1.83 m, 1.71 m | 25.3 CH2 | 1.87 m, 1.75 m |
| 2 | 32.8 CH2 | 3.76 br d (7.5), 2.22 m | 32.9 CH2 | 3.81 br d (7.5), 2.16 m | 33.2 CH2 | 3.87 m, 2.29 m |
| 3 | 217.8 C | 217.6 C | 217.6 C | |||
| 4 | 80.3 C | 80.4 C | 80.3 C | |||
| 5 | 78.2 CH | 5.36 dd (8.5, 2.0) | 78.1 CH | 5.37 dd (9.6, 1.2) | 78.1 CH | 5.39 br d (9.5) |
| 6 | 79.8 CH | 4.60 dd (8.5, 5.0) | 79.9 CH | 4.65 dd (9.6, 6.0) | 79.9 CH | 4.69 dd (9.5,6.5) |
| 7 | 41.5 CH | 2.97 m | 41.6 CH | 3.01 m | 41.7 CH | 3.02 m |
| 8 | 70.3 CH | 4.35 d (10.5) | 70.3 CH | 4.43 d (10.2) | 70.5 CH | 4.40 d (10.0) |
| 9 | 78.7 CH | 5.11 d (10.5) | 79.3 CH | 5.18 d (10.2) | 78.4 CH | 5.15 d (10.0) |
| 10 | 29.8 CH | 2.15 m | 30.1 CH | 2.21 m | 30.0 CH | 2.23 m |
| 11 | 132.6 C | 132.7 C | 132.7 C | |||
| 12 | 169.6 C | 169.6 C | 169.5 C | |||
| 13 | 123.9 CH2 | 6.27 d (3.0), 5.62 d (3.0) | 123.8 CH2 | 6.30 d (3.0) 5.67 d (3.0) | 123.8 CH2 | 6.32 d (3.0) 5.67 d (3.0) |
| 14 | 20.0 CH3 | 0.92 d (7.0) | 19.9 CH3 | 0.94 d (6.6) | 20.0 CH3 | 0.98 d (6.5) |
| 15 | 23.4 CH3 | 1.18 s | 23.3 CH3 | 1.21 s | 23.5 CH3 | 1.24 s |
| 1′ | 172.5 C | 176.3 C | 175.9 C | |||
| 2′ | 42.7 CH2 | 2.31 d (7.0), 2.26 o | 33.9 CH | 2.67 m | 41.3 CH | 2.52 m |
| 3′ | 25.3 CH | 2.09 o c | 18.0 CH3 | 1.21 d (6.6) | 26.3 CH2 | 1.76 o, 1.52 o |
| 4′ | 21.3 CH3 | 0.96 d (6.0) | 17.9 CH3 | 1.20 d (6.6) | 16.1 CH3 | 1.24 d (7.0) |
| 5′ | 21.4 CH3 | 0.95 d (6.0) | 10.7 CH3 | 0.98 t (7.0) | ||
| 1′′ | 173.3 C | 167.2 C | 176.7 C | |||
| 2′′ | 43.0 CH2 | 2.26 o, 2.06 o | 136.4 C | 41.5 CH | 2.52 m | |
| 3′′ | 25.4 CH | 2.09 o | 124.7 CH2 | 5.63 dq (3.6, 1.8), 6.13 dq (3.6, 1.8) | 26.2 CH2 | 1.76 o, 1.52 o |
| 4′′ | 21.4 CH3 | 0.96 d (6.5) | 17.1 CH3 | 1.96 br s | 16.2 CH3 | 1.26 d (7.0) |
| 5′′ | 21.4 CH3 | 0.95 d (6.5) | 10.6 CH3 | 0.96 t (7.0) | ||
| Compounds | IC50 (μM) | ||
|---|---|---|---|
| A549 | HepG2 | Hela | |
| 1 | >40 | 16.98 ± 2.23 | 29.39 ± 0.17 |
| 2 | 30.70 ± 0.51 | 7.47 ± 0.21 | 16.82 ± 0.27 |
| 3 | >40 | >40 | >40 |
| 4 | >40 | >40 | >40 |
| 5 | >40 | 31.64 ± 0.16 | 11.63 ± 1.00 |
| cis-platin | 7.90 ± 0.23 | 13.03 ± 1.49 | 15.34 ± 0.35 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Si, J.-G.; Zhang, Q.-B.; Chen, J.-H.; Ding, G.; Zhang, H.-W.; Jia, H.-M.; Zou, Z.-M. Three New Highly Oxygenated Germacranolides from Carpesium Divaricatum and Their Cytotoxic Activity. Molecules 2018, 23, 1078. https://doi.org/10.3390/molecules23051078
Zhang T, Si J-G, Zhang Q-B, Chen J-H, Ding G, Zhang H-W, Jia H-M, Zou Z-M. Three New Highly Oxygenated Germacranolides from Carpesium Divaricatum and Their Cytotoxic Activity. Molecules. 2018; 23(5):1078. https://doi.org/10.3390/molecules23051078
Chicago/Turabian StyleZhang, Tao, Jin-Guang Si, Qiu-Bo Zhang, Jia-Huan Chen, Gang Ding, Hong-Wu Zhang, Hong-Mei Jia, and Zhong-Mei Zou. 2018. "Three New Highly Oxygenated Germacranolides from Carpesium Divaricatum and Their Cytotoxic Activity" Molecules 23, no. 5: 1078. https://doi.org/10.3390/molecules23051078