
Simultaneous Analysis of Iridoid Glycosides and Anthraquinones in Morinda officinalis Using UPLC-QqQ-MS/MS and UPLC-Q/TOF-MSE


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Optimization of UPLC-QqQ-MS/MS Conditions
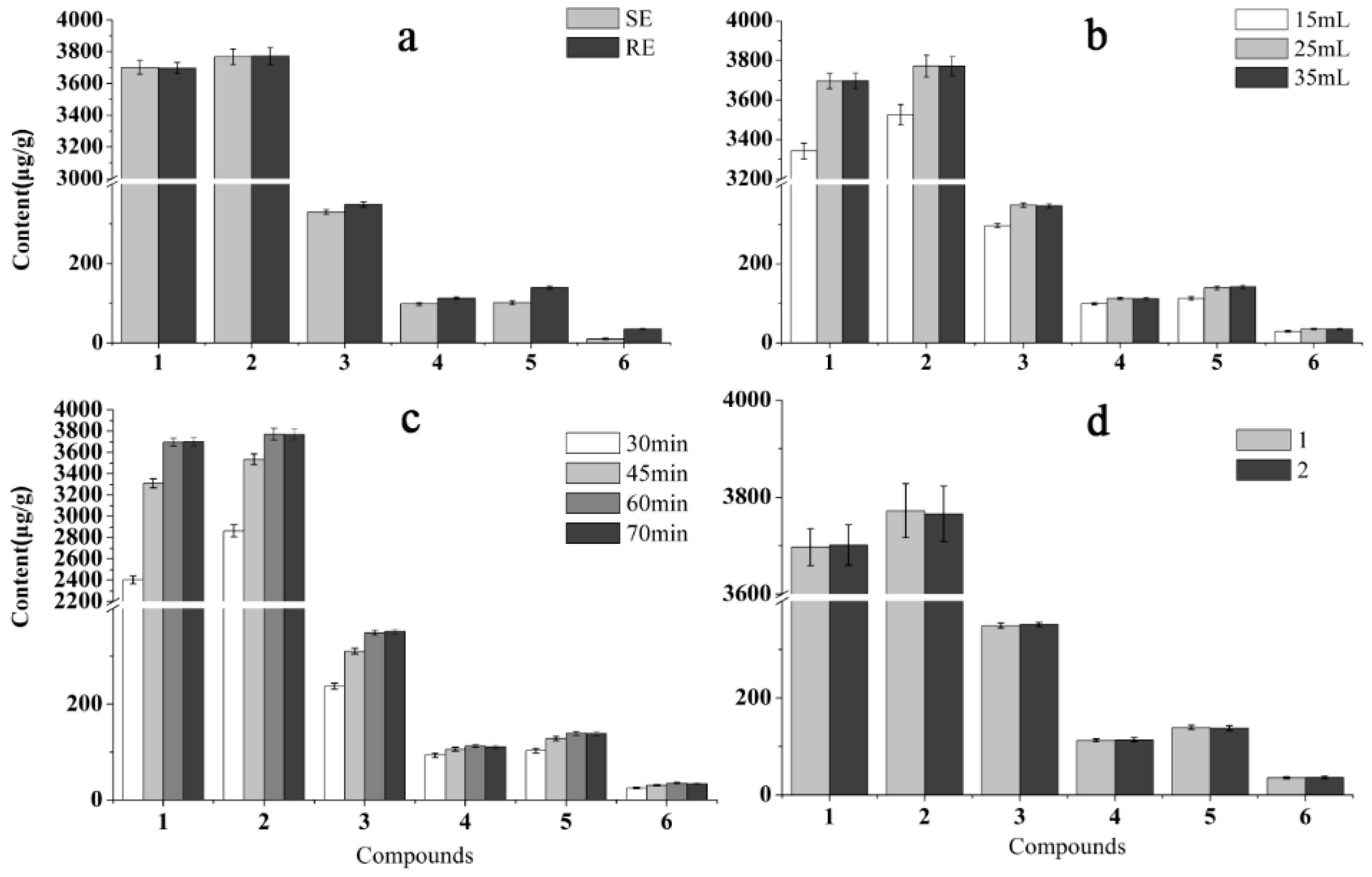
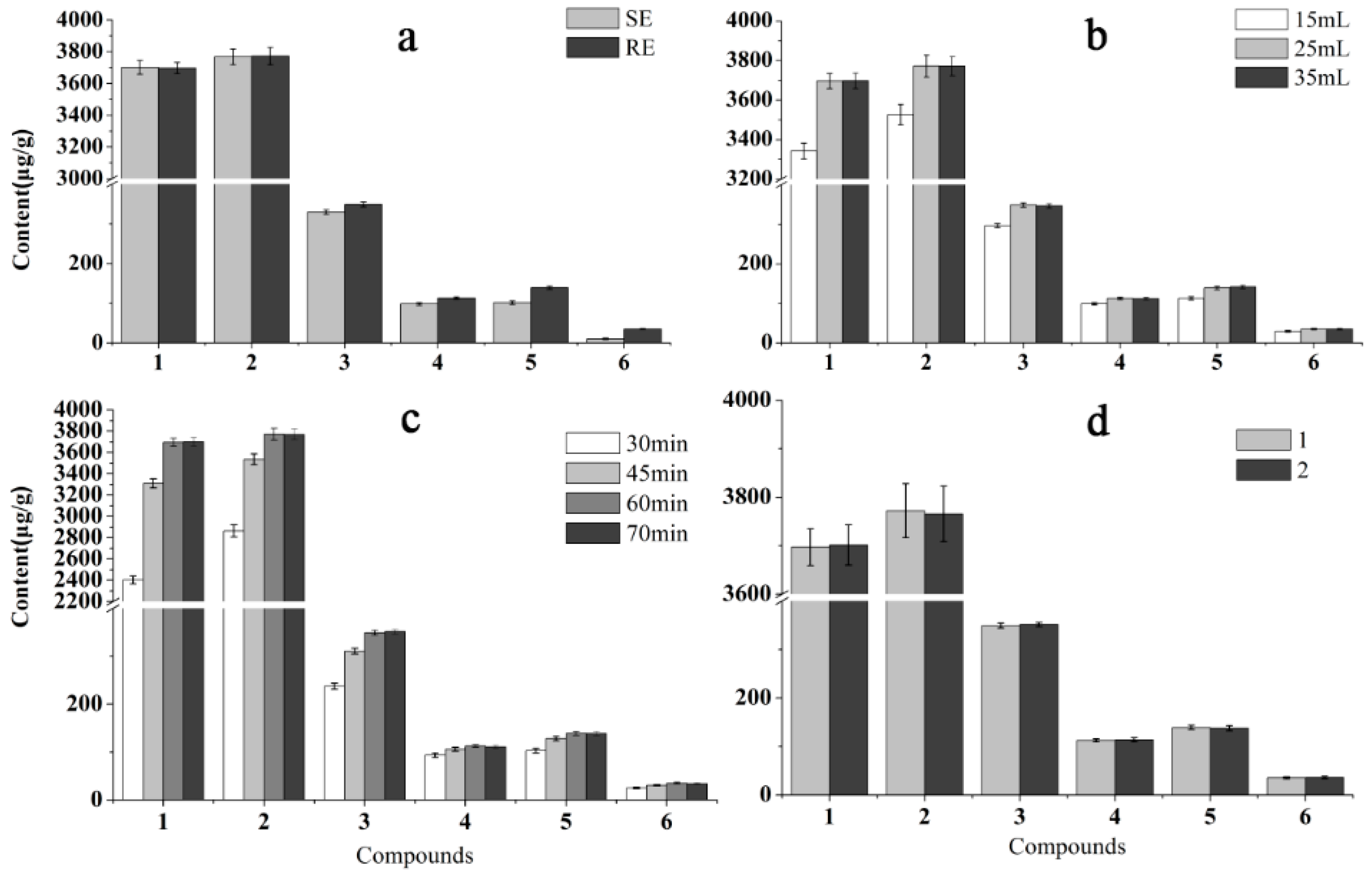
2.2. Optimization of Sample Preparation
2.3. Method Validation
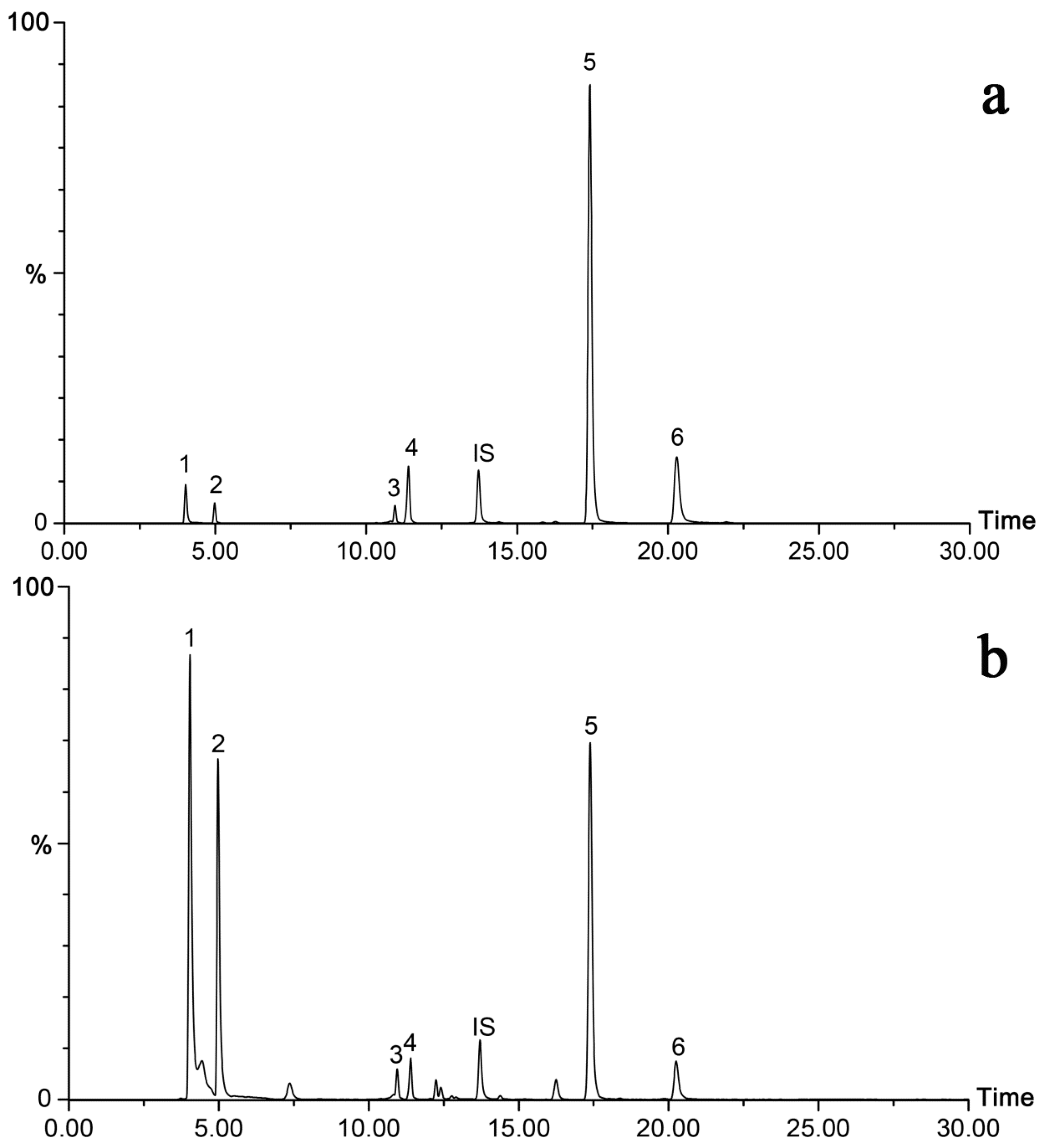
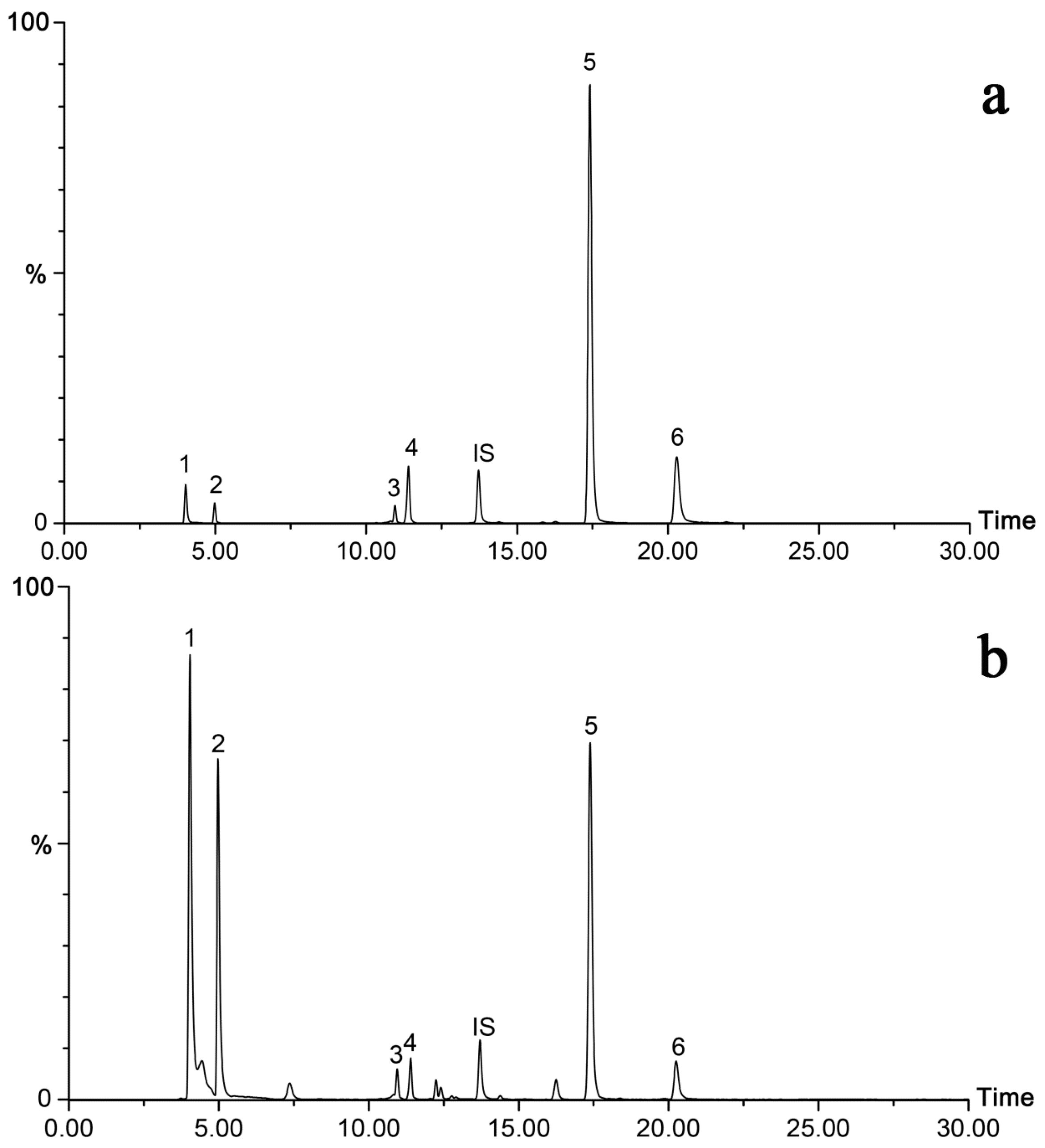
2.4. Sample Analysis
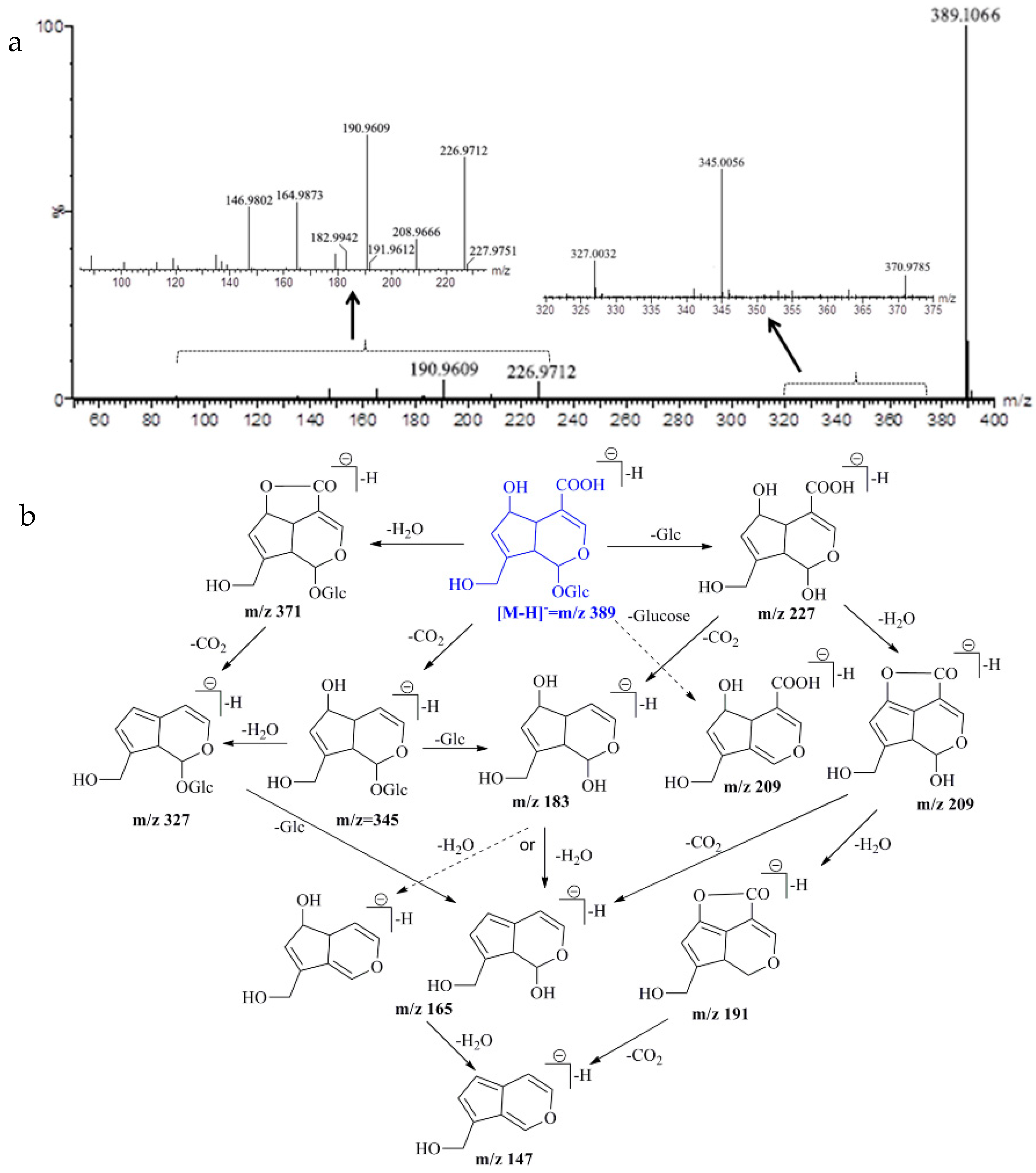
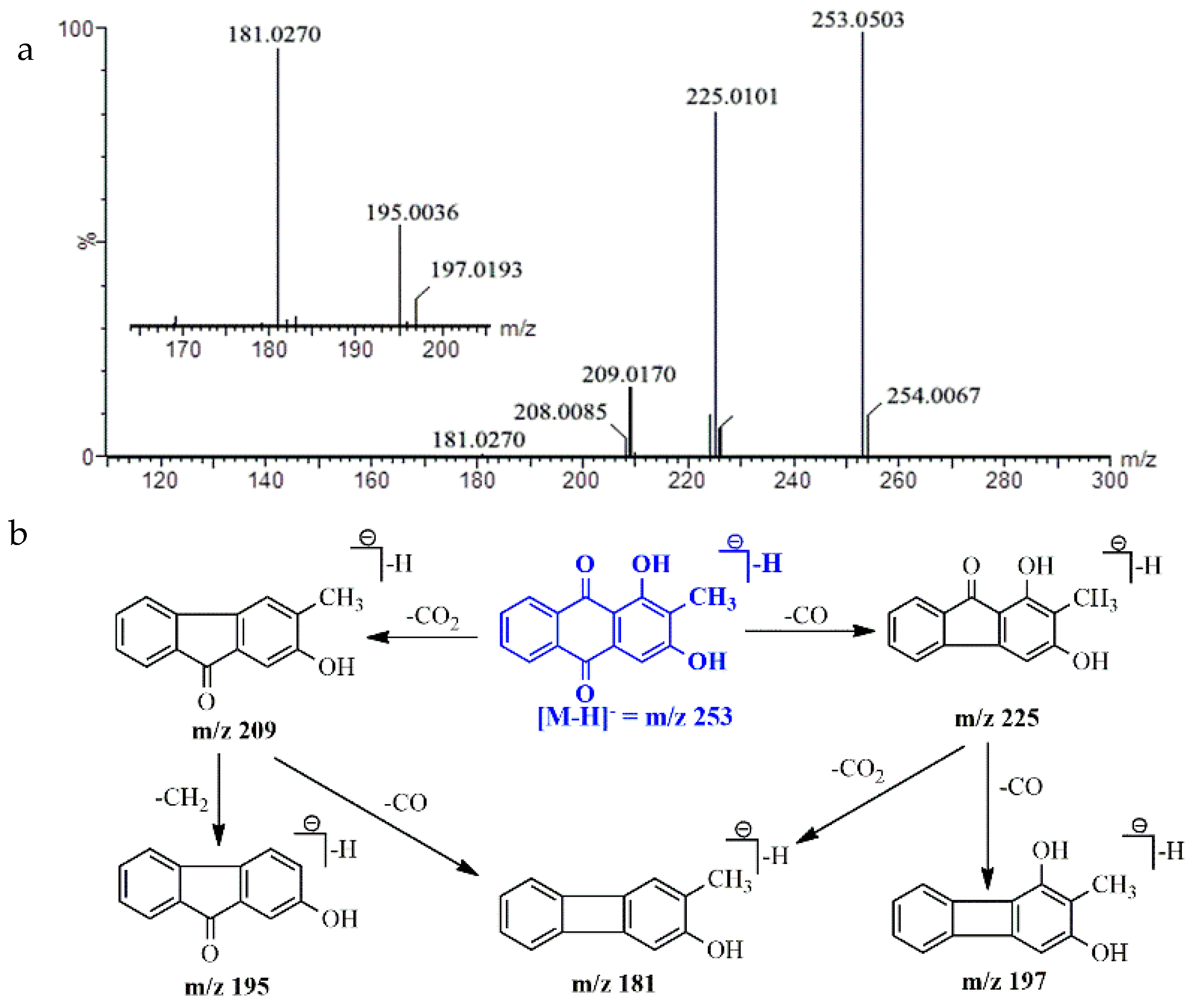
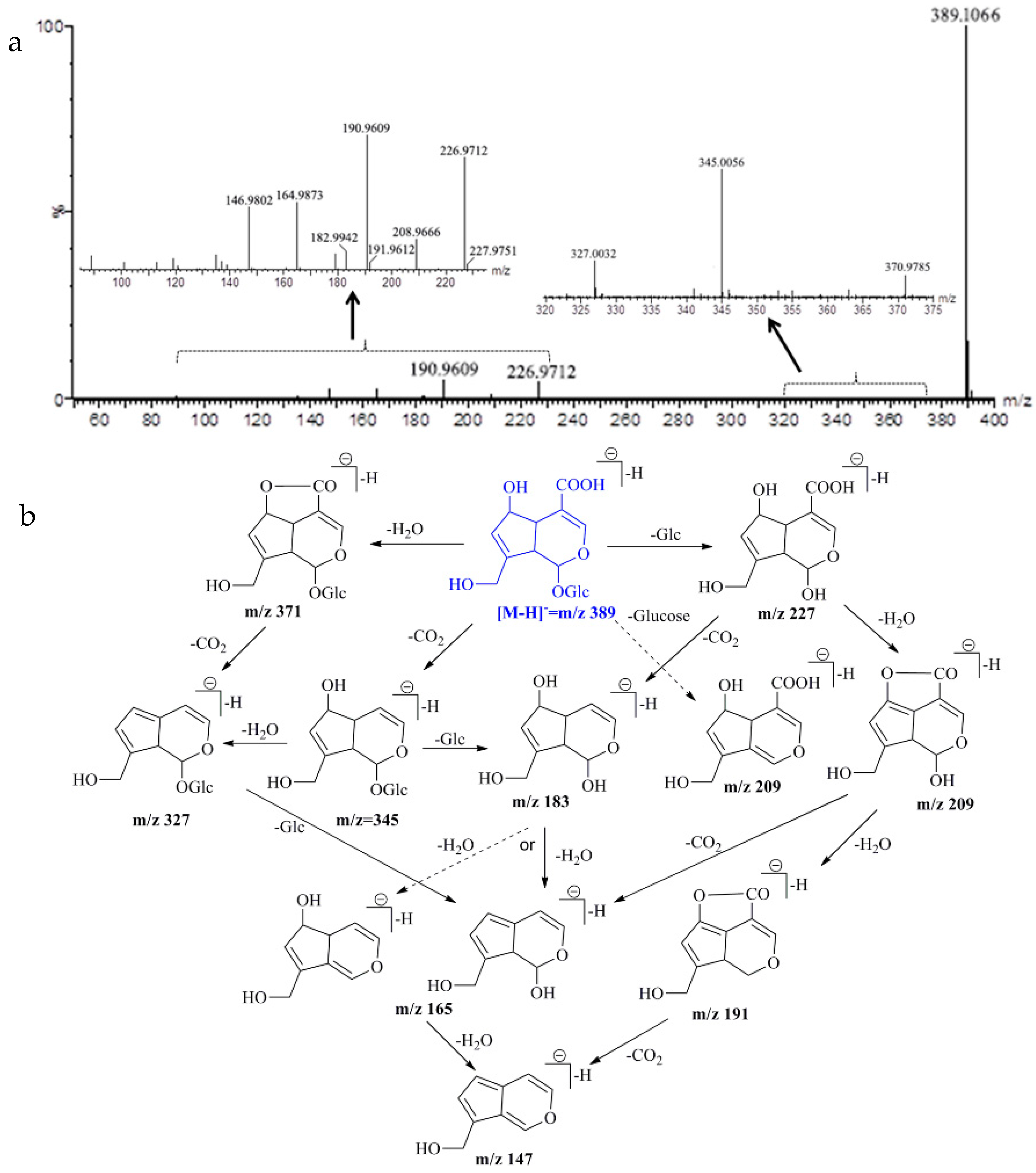
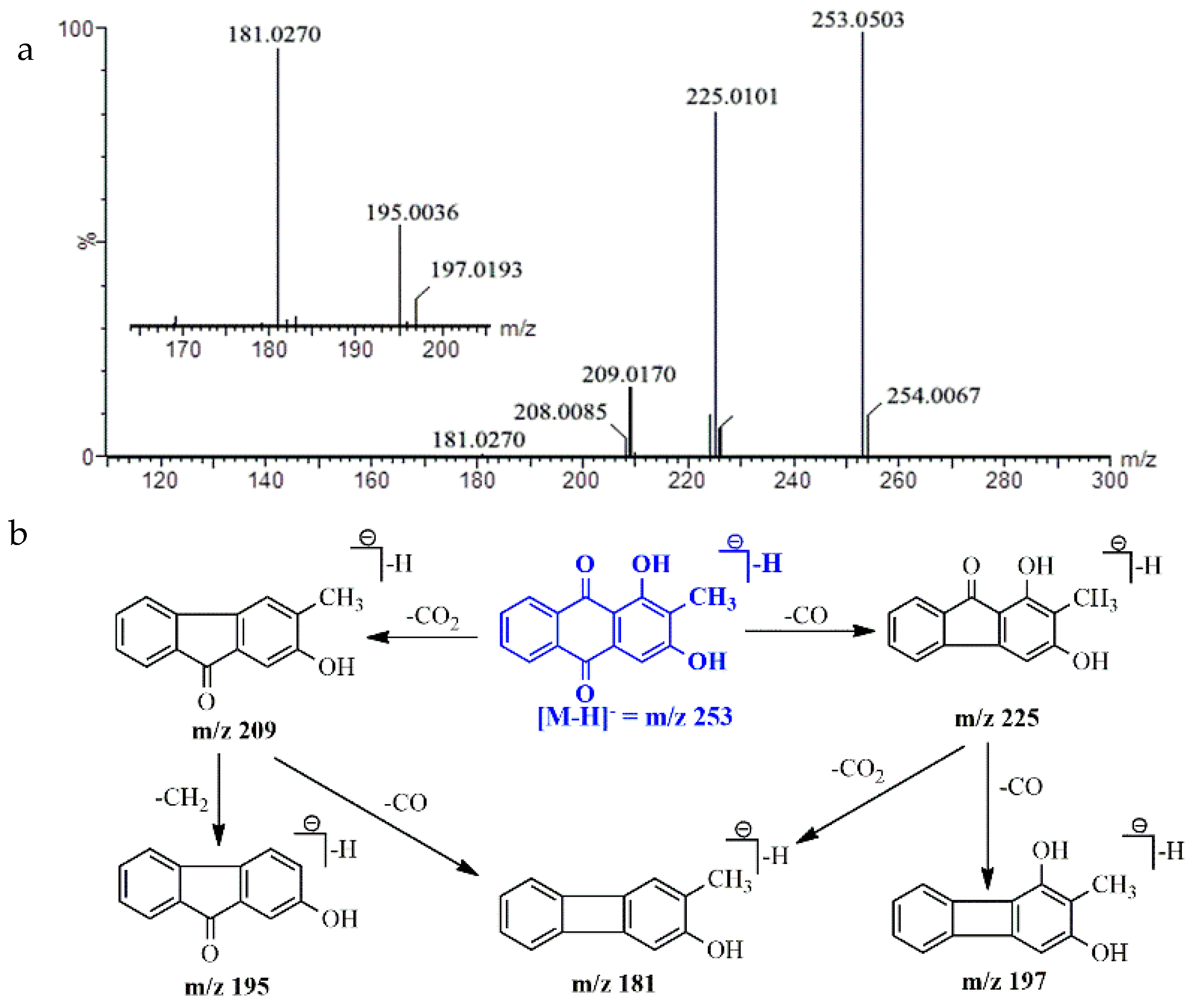
2.5. UPLC-Q/TOF-MS Identification
3. Materials and Methods
3.1. Chemicals, Reagents, and Samples
3.2. UPLC-QqQ-MS/MS
3.3. UPLC-Q/TOF-MS/MS
3.4. Sample Preparation
3.5. Standard Solution Preparation
3.6. Method Validation
3.6.1. Linearity and Limit of Quantification
3.6.2. Precision, Repeatability, and Stability
3.6.3. Recovery
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Yong, J.P.; Lu, C.Z.; Huang, S.J.; Wu, X.Y. Chemical components isolated from the roots of Morinda officinalis. Chem. Nat. Compd. 2015, 51, 548–549. [Google Scholar] [CrossRef]
- Chen, D.L.; Li, N.; Lin, N.; Long, H.M.; Li, H.; Chen, J.; Zhang, H.M.; Zeng, C.C.; Liu, S.H. Confocal mirco-raman spectroscopic analysis of the antioxidant protection mechanism of the oligosaccharides extracted from Morinda officinalis on human sperm DNA. J. Ethnopharmacol. 2014, 153, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Chinese Pharmacopoeia Commission. Pharmacopoeia of People’s Republic of China; Chemical Industry Press: Beijing, China, 2015. [Google Scholar]
- Wu, Y.B.; Zheng, C.J.; Qin, L.P.; Sun, L.N.; Han, T.; Jiao, L.; Zhang, Q.Y.; Zhong, J.Z. Antiosteoporotic activity of anthraquinones from Morinda officinalis on osteoblasts and osteoclasts. Molecules 2009, 14, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Li, J.; Li, G.; Wang, D.M.; Zhu, L.P.; Yang, D.P. Structural characterization and anti–fatigue activity polysaccharides from the roots of Morinda officinalis. Int. J. Biol. Macromol. 2009, 44, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Soon, Y.Y.; Tan, B.K.H. Evaluation of the hypoglycemic and antioxidant activities of Morinda officinalis in streptozotocin—Induced diabetic rats. Singap. Med. J. 2002, 43, 77–85. [Google Scholar]
- Zhang, Z.Q.; Yuan, L.; Yang, M.; Luo, Z.P.; Zhao, Y.M. The effect of Morinda officinalis How, a Chinese traditional medicinal plant, on the DRL 72–s schedule in rats and the forced swimming test in mice. Pharmacol. Biochem. Behav. 2002, 72, 39–43. [Google Scholar] [CrossRef]
- Choi, J.; Lee, K.; Choi, M.; Nam, J.H.; Jung, H.J.; Park, S.K.; Park, H.J. Antinociceptive anti-inflammatory effect of monotropein isolated from the root of Morinda officinalis. Biol. Pharm. Bull. 2005, 28, 1915–1918. [Google Scholar] [CrossRef] [PubMed]
- Li, C.M.; Dong, J.; Tian, J.C.; Deng, Z.P.; Song, X.J. LC/MS/MS determination and pharmacokinetic study of iridoid glycosides monotropein and deacetylasperulosidic acid isomers in rat plasma after oral administration of Morinda officinalis extract. Biomed. Chromatogr. 2016, 30, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Qi, L.W.; Liu, E.H.; Zhou, J.L.; Wen, X.D. Analysis of Chinese herbal medicines with holistic approaches and integrated evaluation models. Trac-Trend Anal. Chem. 2008, 27, 66–77. [Google Scholar] [CrossRef]
- Bao, L.; Qin, L.; Liu, L.; Wu, Y.; Han, T.; Xue, L.; Zhang, Q. Anthraquinone compounds from Morinda officinalis inhibit osteoclastic bone resorption in vitro. Chem.-Biol. Interact. 2011, 194, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Zhang, Q.Y.; Yang, H.; Liu, W.; Zhang, N.D.; Qiu, L.P.; Xin, H.L. Monotropein isolated from the roots of Morinda officinalis increases osteoblastic bone formation and prevents bone loss in ovariectomized mice. Fitoterapia 2016, 110, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Kyoung, K.; Byungkil, C.; Mi, C.; Yeong, L.; Won, L. Separation and quantitative analysis of anthraquinones in Morinda officinalis How. by HPLC. Planta Med. 2006, 72, 1069. [Google Scholar] [CrossRef]
- Shi, J.; Liu, Z.; Wang, L.; Ji, T.Z. Determination of five active components in Morinda officinalis from different habitats by HPLC. Chin. Med. Mat. 2015, 38, 245–248. [Google Scholar]
- Wang, Y.L.; Cui, H.M.; Huang, S.J.; Li, Q.; Lei, H.M. Determination of main iridoid glycosides from Morindae officinalis Radix of different producting areas by HPLC. Chin. Med. Mat. 2011, 34, 1187–1190. [Google Scholar]
- Yang, T.C.; Xu, J.; Liang, Y.; Liu, J.; Ding, P. Determination of momotropein from Radix Morindae officinalis by HPLC. West China J. Pharm. Sci. 2008, 23, 695–696. [Google Scholar]
- Zhao, X.S.; Kong, W.J.; Zhou, Y.K.; Wei, J.H.; Yang, M.H. Evaluation and quantitative analysis of 11 compounds in Morinda officinalis using ultra-performance liquid chromatography-photodiode array coupled with chemometrics. J. Sep. Sci. 2017, 40, 3996–4003. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, L.; Duan, L.; Dong, X.; Zhou, P.; Liu, E.H.; Li, P. Simultaneous determination of 16 phenolic constituents in Spatholobi caulis by high performance liquid chromatography/electrospray ionization triple quadrupole mass spectrometry. J. Pharm. Biomed. Anal. 2015, 102, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Chandra, P.; Bajpai, V.; Singh, A.; Srivastava, M.; Mishra, D.K.; Kumar, B. Rapid qualitative and quantitative analysis of bioactive compounds from Phyllanthus amarus using LC/MS/MS techniques. Ind. Crop Prod. 2015, 69, 143–152. [Google Scholar] [CrossRef]
- Wu, Y.H.; Jiang, X.L.; Zhang, S.X.; Dai, X.L.; Liu, Y.J.; Tan, H.R.; Gao, L.P.; Xia, T. Quantification of flavonol glycosides in Camellia sinensis by MRM mode of UPLC-QQQ-MS/MS. J. Chromatogr. B 2016, 1017–1018, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.F.; Wang, Y.W.; Ouyang, H.; Lu, Y.; Qiu, Y.; Feng, Y.L.; Jiang, H.L.; Zhou, X.; Yang, S.L. A novel dereplication strategy for the identification of two new trace compounds in the extract of Gastrodia elata using UHPLC-Q/TOF-MS/MS. J. Chromatogr. B 2015, 988, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.F.; Jiao, Z.; Liu, Y.; Li, Z.D.; Shi, X.J.; Wang, W.J.; Wang, B.; Zhong, M.K. Chemical profiling of San-Huang decoction by UPLC–ESI-Q-TOF-MS. J. Pharm. Biomed. Anal. 2016, 131, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Deng, H.Z.; Mo, Q.L.; Lin, Z.W. Determination of the contents of valuable compositions of Morinda officinalis How in different regions and analysis of its protection for the liver. J. Hainan Norm. Univ. 2003, 16, 64–67. [Google Scholar]
- Xu, J.Y.; Liang, Y.J.; Ding, P. Variations of monotropein in Morinda officinalis collected in different growth period. Chin. Tradit. Herb. Drugs 2007, 38, 772–774. [Google Scholar]
- Li, C.M.; Zhang, X.L.; Xue, X.Y.; Zhang, F.Y.; Xu, Q.; Liang, X.M. Structural characterization of iridoid glycosides by ultra-performance liquid chromatography/electrospray ionization quadrupole time-of-flight tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 1941–1954. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Wang, H.; Wan, L.R.; Hashi, Y.; Chen, S.Z. Identification and determination of major constituents in Polygonum cuspidatum Sieb. et Zucc by high performance liquid chromatography/electrospray ionization-ion trap/time-of-flight mass spectrometry. Chin. J. Chromatogr. 2009, 27, 425–430. [Google Scholar]
- Wang, M.L.; Zhang, Q.Q.; Fu, S.; Liu, Y.H.; Liang, C.L.; Chen, N.; Ding, Y.S.; Wang, H.L.; Qu, X.M.; Zhao, B.S.; et al. Characterization of Morinda officinalis How. by UPLC-Q-TOF-MSE coupled with UNIFI database filiter. J. Chin. Mass Spectrom. Soc. 2017, 38, 75–82. [Google Scholar]
- Li, C.M.; Xue, X.Y.; Zhou, D.Y.; Zhang, F.F.; Xu, Q.; Ren, L.L.; Liang, X.M. Analysis of iridoid glycosides in Hedyotis diffusa by high-performance liquid chromatography/electrospray ionization tandem mass spectrometry. J. Pharm. Biomed. Anal. 2008, 48, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhang, L.; Wang, W.H.; Liu, B. Components identification in Cassiae Semen by HPLC-IT-TOF-MS. J. Pharm. Anal. 2015, 35, 1408–1411. [Google Scholar]
- Dong, H.J.; Chen, X.H.; Zeng, R. Rapid analysis on chemical constituents in roots of Rheum pumilum by UPLC coupled with hybrid quadrupole-orbit trap MS. Chin. Tradit. Herb. Drugs 2016, 47, 2428–2435. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | Retention Time (min) | Parent (m/z) | Daughter (m/z) (Q/I) a | Cone Voltage (V) | Collision Energy (eV) |
|---|---|---|---|---|---|
| Monotropein | 4.03 | 388.98 | 146.95/190.95 | 47 | 20/25 |
| Deacetyl asperulosidic acid | 4.98 | 389.12 | 118.93/164.95 | 45 | 24/21 |
| Asperulosidic acid | 10.96 | 431.05 | 251.03/165.01 | 48 | 22/20 |
| Asperuloside | 11.41 | 413.03 | 146.95/190.94 | 51 | 18/23 |
| Rubiadin-1-methyl ether | 17.42 | 267.02 | 223.99/251.95 | 50 | 25/25 |
| Rubiadin | 20.29 | 252.97 | 224.01/209.96 | 50 | 30/26 |
| Jaceosidin (IS) | 13.73 | 328.98 | 313.95/298.96 | 50 | 22/22 |
| Analytes | Calibration Curves | R2 | Linear Range (μg/mL) | Precision (RSD, %) | LOQ ng/mL | LOD ng/mL | Repeatability (RSD, %, n = 6) | |
|---|---|---|---|---|---|---|---|---|
| Intra-Day | Inter-Day | |||||||
| Monotropein | Y = 0.0492X + 0.0072 | 0.9981 | 0.015–79.65 | 1.71 | 3.80 | 13.87 | 4.20 | 2.89 |
| Deacetyl asperulosidic acid | Y = 0.0281X + 0.001 | 0.9990 | 0.030–96.00 | 2.80 | 3.83 | 26.63 | 8.07 | 3.92 |
| Asperulosidic acid | Y = 0.0252X + 0.0018 | 0.9956 | 0.042–21.00 | 1.12 | 2.56 | 27.57 | 9.19 | 2.54 |
| Asperuloside | Y = 0.1235X + 0.0073 | 0.9987 | 0.018–37.50 | 2.71 | 2.74 | 11.06 | 3.35 | 3.17 |
| Rubiadin-1-methyl ether | Y = 1.1937 X + 0.2136 | 0.9930 | 0.008–21.75 | 2.50 | 4.21 | 6.74 | 2.04 | 4.02 |
| Rubiadin | Y = 0.587X + 0.0233 | 0.9949 | 0.003–22.50 | 0.75 | 2.72 | 2.60 | 0.87 | 2.35 |
| Analytes | Sample (g) | Origin (μg) | Spiked (μg) | Found (μg) | Mean Recovery (%) (RSD, %) |
|---|---|---|---|---|---|
| Monotropein | 0.5 | 1848.45 | 1480 | 3304.33 | 98.37 (3.23) |
| 1850 | 3680.69 | 99.04 (2.15) | |||
| 2220 | 4059.13 | 99.58 (3.06) | |||
| Deacetyl asperulosidic acid | 0.5 | 1883.74 | 1500 | 3351.19 | 97.83 (4.07) |
| 1880 | 3731.03 | 98.26 (2.38) | |||
| 2260 | 4113.01 | 98.64 (1.95) | |||
| Asperulosidic acid | 0.5 | 174.43 | 140 | 310.70 | 97.33 (2.84) |
| 175 | 346.00 | 98.04 (3.37) | |||
| 210 | 384.14 | 99.86 (3.69) | |||
| Asperuloside | 0.5 | 56.32 | 45 | 100.61 | 98.41 (4.23) |
| 55 | 110.13 | 97.83 (3.94) | |||
| 67.5 | 122.63 | 98.23 (2.78) | |||
| Rubiadin-1-methyl ether | 0.5 | 69.53 | 55 | 122.58 | 96.47 (2.94) |
| 70 | 136.56 | 95.77 (2.62) | |||
| 84 | 149.59 | 95.32 (3.15) | |||
| Rubiadin | 0.5 | 17.62 | 14 | 31.27 | 97.48 (1.75) |
| 17.5 | 34.63 | 97.17 (2.77) | |||
| 21 | 37.88 | 96.48 (3.28) |
| No. | Origins | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|---|
| S1 | Guangdong | 2497.58 | 2766.00 | 246.94 | 33.95 | 113.49 | 11.15 |
| S2 | Guangdong | 2125.89 | 2179.01 | 171.42 | 8.44 | 184.22 | 21.64 |
| S3 | Guangdong | 1861.98 | 1771.73 | 83.22 | ND | 116.76 | 7.95 |
| S4 | Guangdong | 2141.90 | 2162.22 | 175.05 | 5.98 | 107.61 | 15.67 |
| S5 | Guangdong | 1016.25 | 817.64 | ND | ND | 42.93 | 3.80 |
| S6 | Guangdong | 1590.15 | 1510.59 | 93.76 | 7.38 | 57.04 | 19.31 |
| S7 | Guangdong | 1540.68 | 1593.63 | 46.14 | 1.09 | 146.63 | 17.58 |
| S8 | Guangdong | 1513.27 | 1458.78 | 67.97 | 6.04 | 143.97 | 208.70 |
| S9 | Guangdong | 1827.98 | 1695.86 | 86.50 | 3.08 | 87.69 | 12.79 |
| S10 | Hainan | 2310.95 | 1990.72 | 91.30 | 15.19 | 81.43 | 28.17 |
| S11 | Hainan | 1907.49 | 1697.96 | 151.21 | 15.03 | 118.81 | 38.88 |
| S12 | Hainan | 3020.44 | 3308.44 | 234.72 | 19.95 | 65.85 | 7.01 |
| S13 | Hainan | 3696.90 | 3767.48 | 348.87 | 112.64 | 139.05 | 35.25 |
| S14 | Guangxi | 2046.66 | 1838.41 | 50.22 | 1.13 | 116.43 | 10.84 |
| S15 | Guangxi | 1521.74 | 1784.73 | 116.21 | 11.25 | 167.85 | 24.15 |
| S16 | Fujian | 1940.38 | 1805.78 | 33.05 | 2.07 | 172.22 | 20.89 |
| S17 | Fujian | 1266.58 | 1199.37 | ND | ND | 123.04 | 11.40 |
| Analyte | Molecular Formula | Theoretical Mass (Da) | Measured Mass (Da) | Error (ppm) | Fragment Ions (ESI−, m/z) |
|---|---|---|---|---|---|
| Monotropein | C16H22O11 | 389.1084 [M − H]− | 389.1066 [M − H]− | −4.62 | 226.9712, 190.9609, 164.9873, 146.9802 |
| Deacetyl asperulosidic acid | C16H22O11 | 389.1084 [M − H]− | 389.1072 [M − H]− | −3.08 | 146.9305, 165.0189, 227.0082, 190.9921 |
| Asperulosidic acid | C18H24O12 | 431.1190 [M − H]− | 431.1184 [M − H]− | −1.39 | 146.9305, 165.0189, 251.0062, 119.0033 |
| Asperuloside | C18H22O11 | 413.1084 [M − H]− | 413.1092 [M − H]− | 1.94 | 146.9926, 190.9722, 233.0963, 369.1387 |
| Rubiadin-1-methyl ether | C16H12O4 | 267.0657 [M − H]− | 267.0653 [M − H]− | 1.50 | 224.0036, 252.9935 |
| Rubiadin | C15H10O4 | 253.0501 [M − H]− | 253.0503 [M − H]− | 0.79 | 225.0101, 209.0170, 181.0270 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Wei, J.; Yang, M. Simultaneous Analysis of Iridoid Glycosides and Anthraquinones in Morinda officinalis Using UPLC-QqQ-MS/MS and UPLC-Q/TOF-MSE. Molecules 2018, 23, 1070. https://doi.org/10.3390/molecules23051070
Zhao X, Wei J, Yang M. Simultaneous Analysis of Iridoid Glycosides and Anthraquinones in Morinda officinalis Using UPLC-QqQ-MS/MS and UPLC-Q/TOF-MSE. Molecules. 2018; 23(5):1070. https://doi.org/10.3390/molecules23051070
Chicago/Turabian StyleZhao, Xiangsheng, Jianhe Wei, and Meihua Yang. 2018. "Simultaneous Analysis of Iridoid Glycosides and Anthraquinones in Morinda officinalis Using UPLC-QqQ-MS/MS and UPLC-Q/TOF-MSE" Molecules 23, no. 5: 1070. https://doi.org/10.3390/molecules23051070
APA StyleZhao, X., Wei, J., & Yang, M. (2018). Simultaneous Analysis of Iridoid Glycosides and Anthraquinones in Morinda officinalis Using UPLC-QqQ-MS/MS and UPLC-Q/TOF-MSE. Molecules, 23(5), 1070. https://doi.org/10.3390/molecules23051070