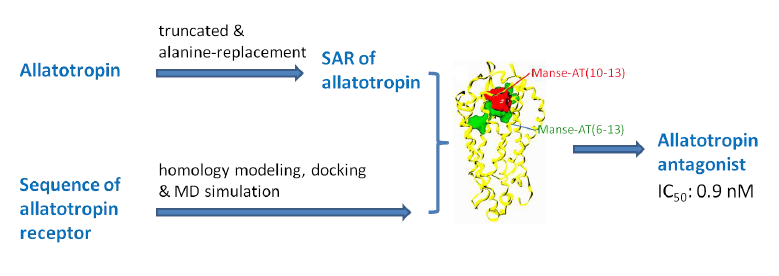
Discovery of a Manduca sexta Allatotropin Antagonist from a Manduca sexta Allatotropin Receptor Homology Model


Abstract
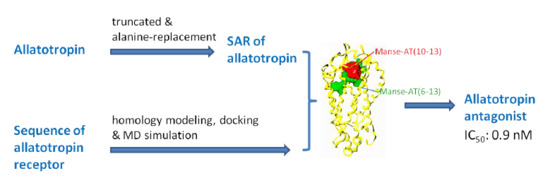
:
1. Introduction
2. Results and Discussion
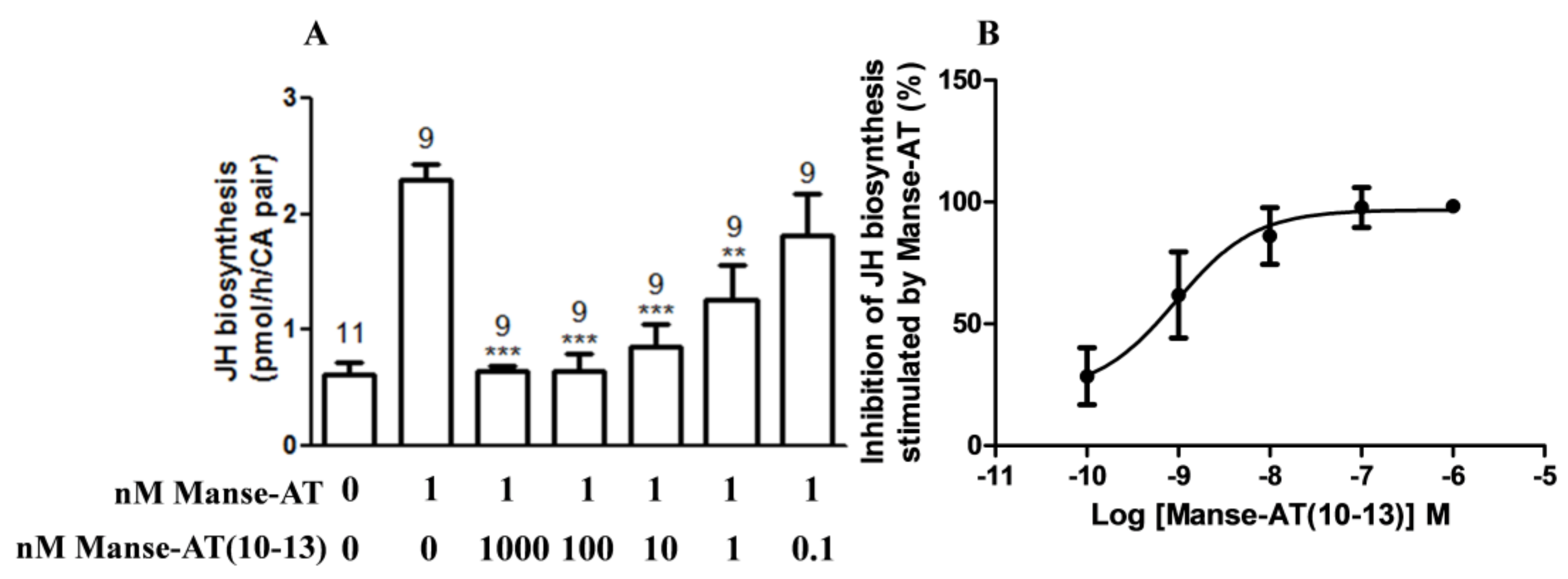
2.1. Effects of Manse-AT and Analogs on JH Biosynthesis
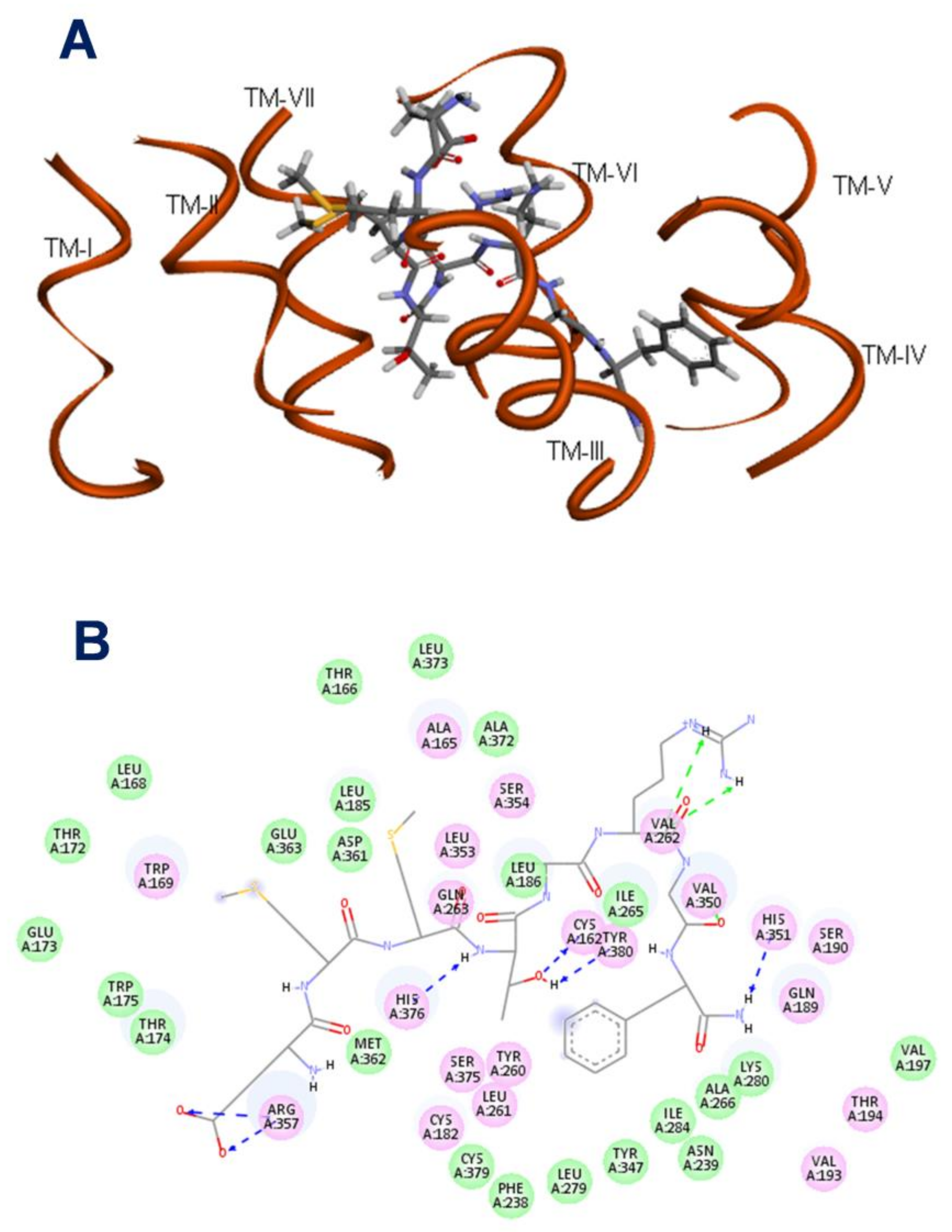
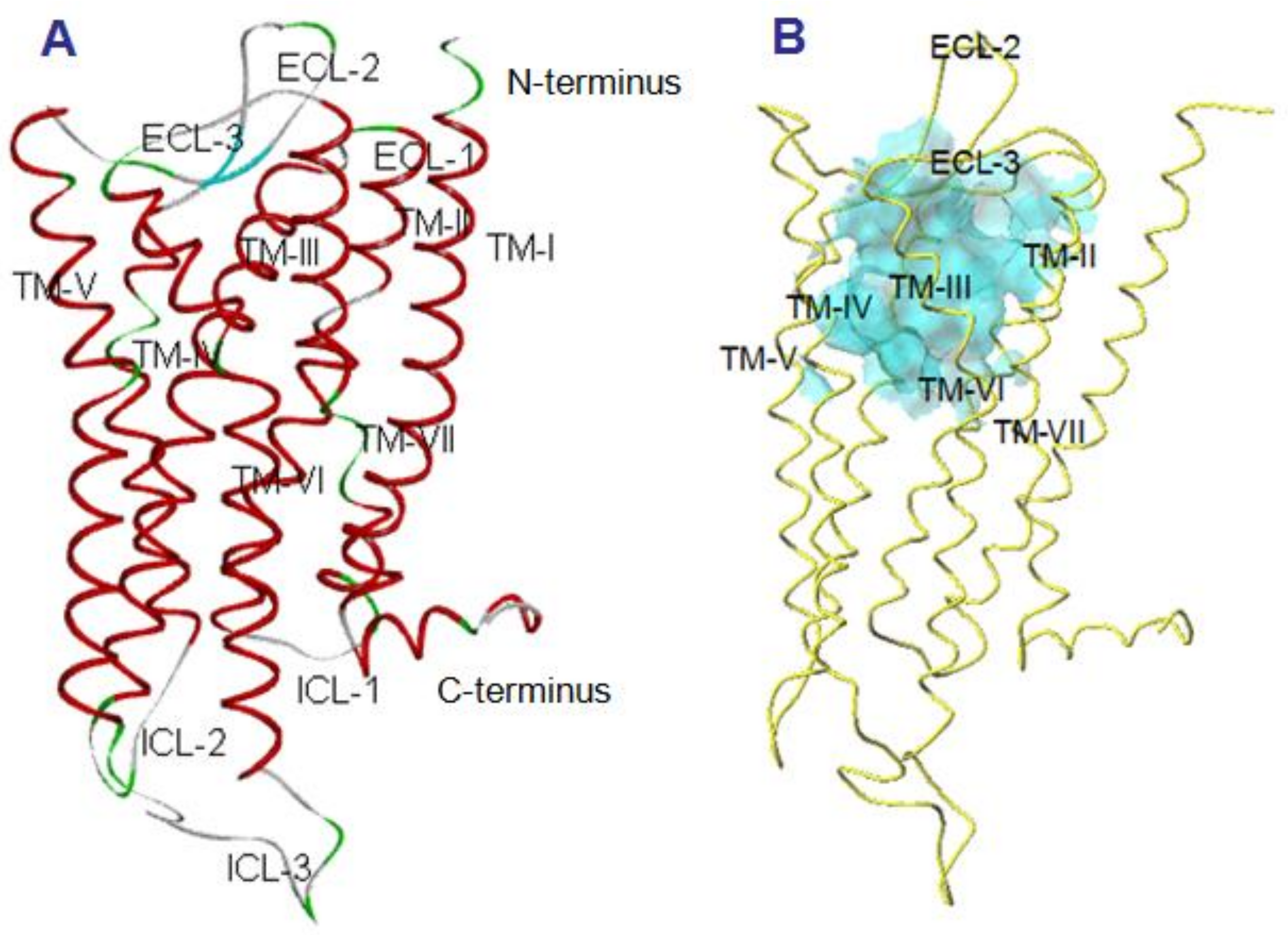
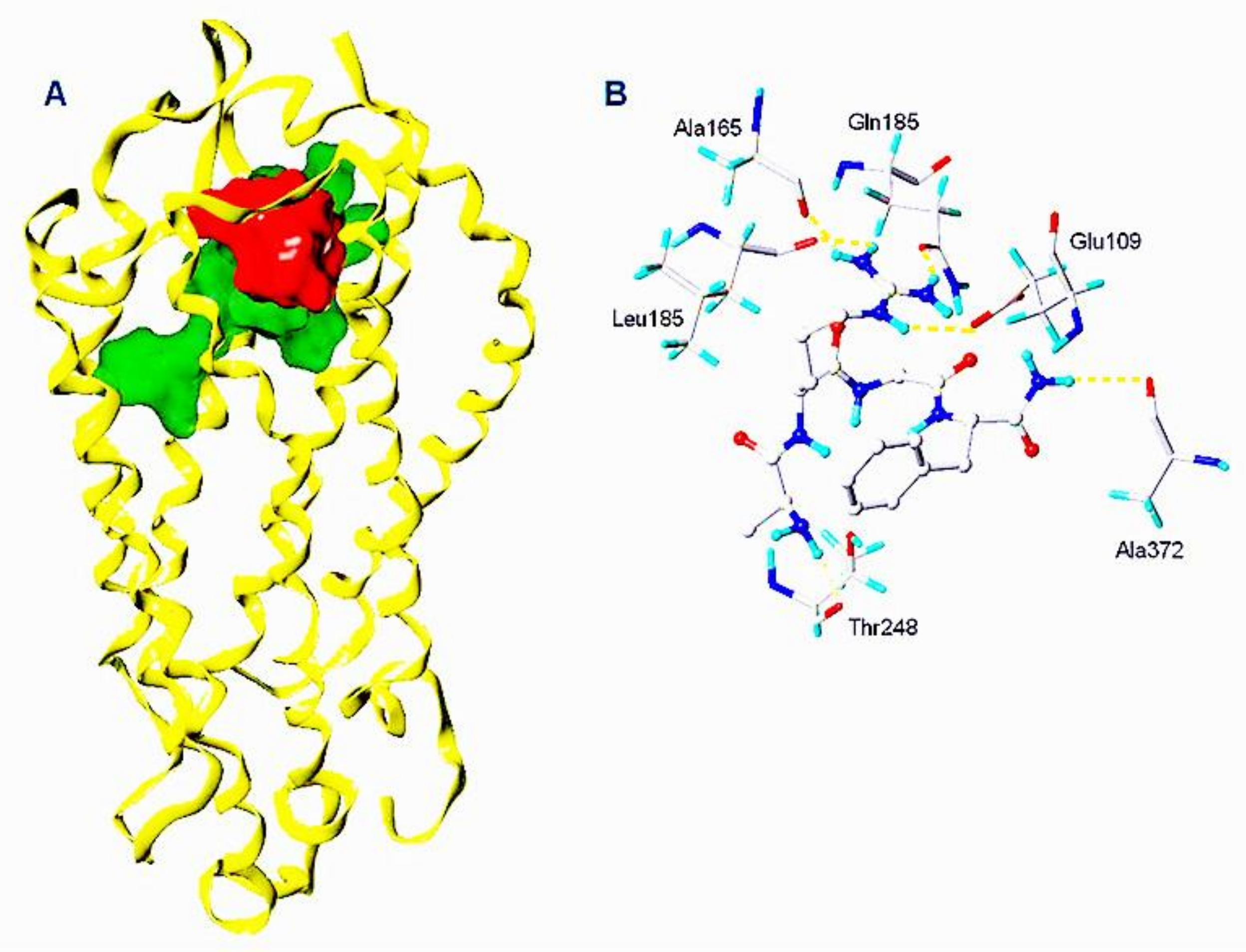
2.2. Manse-ATR Model
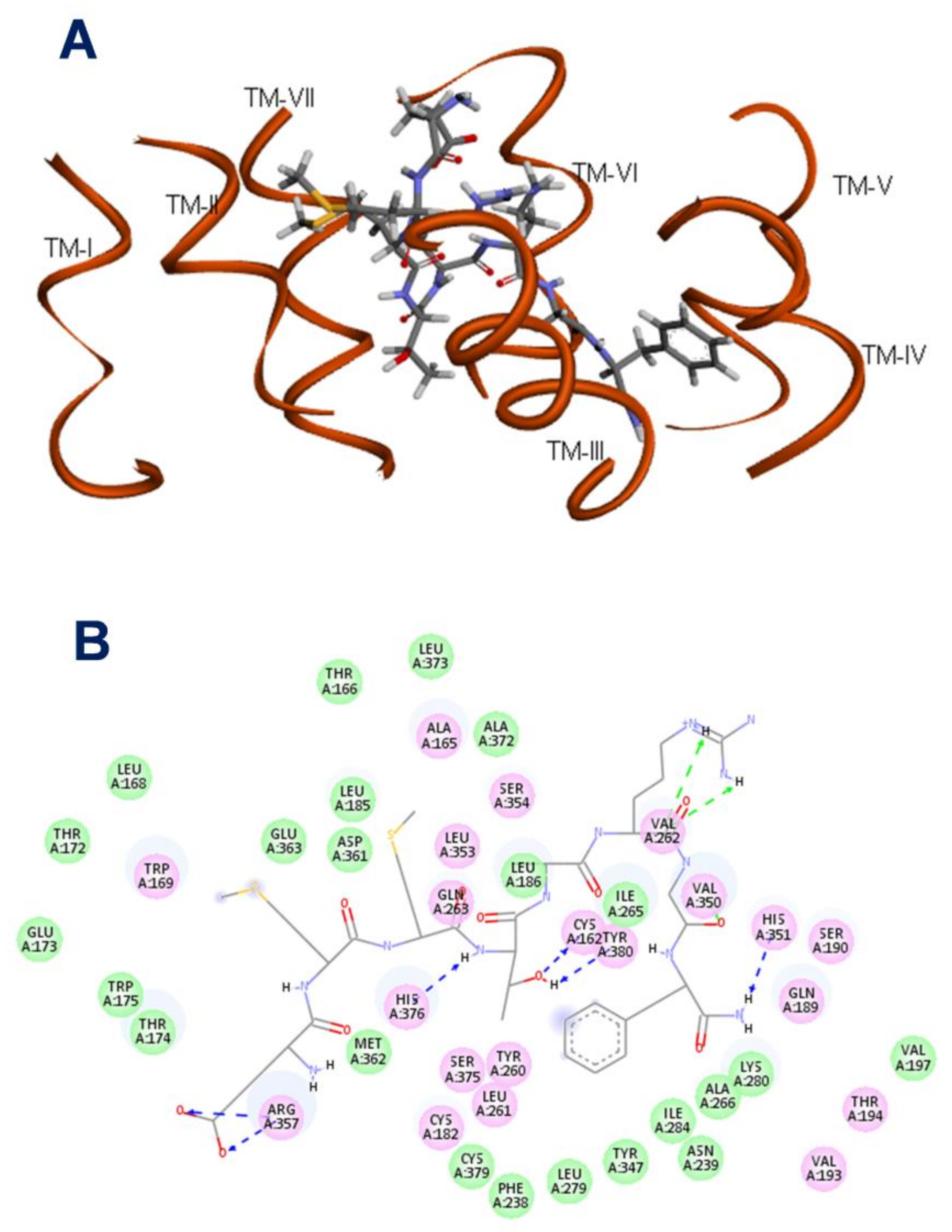
2.3. The Binding Pocket in Manse-ATR
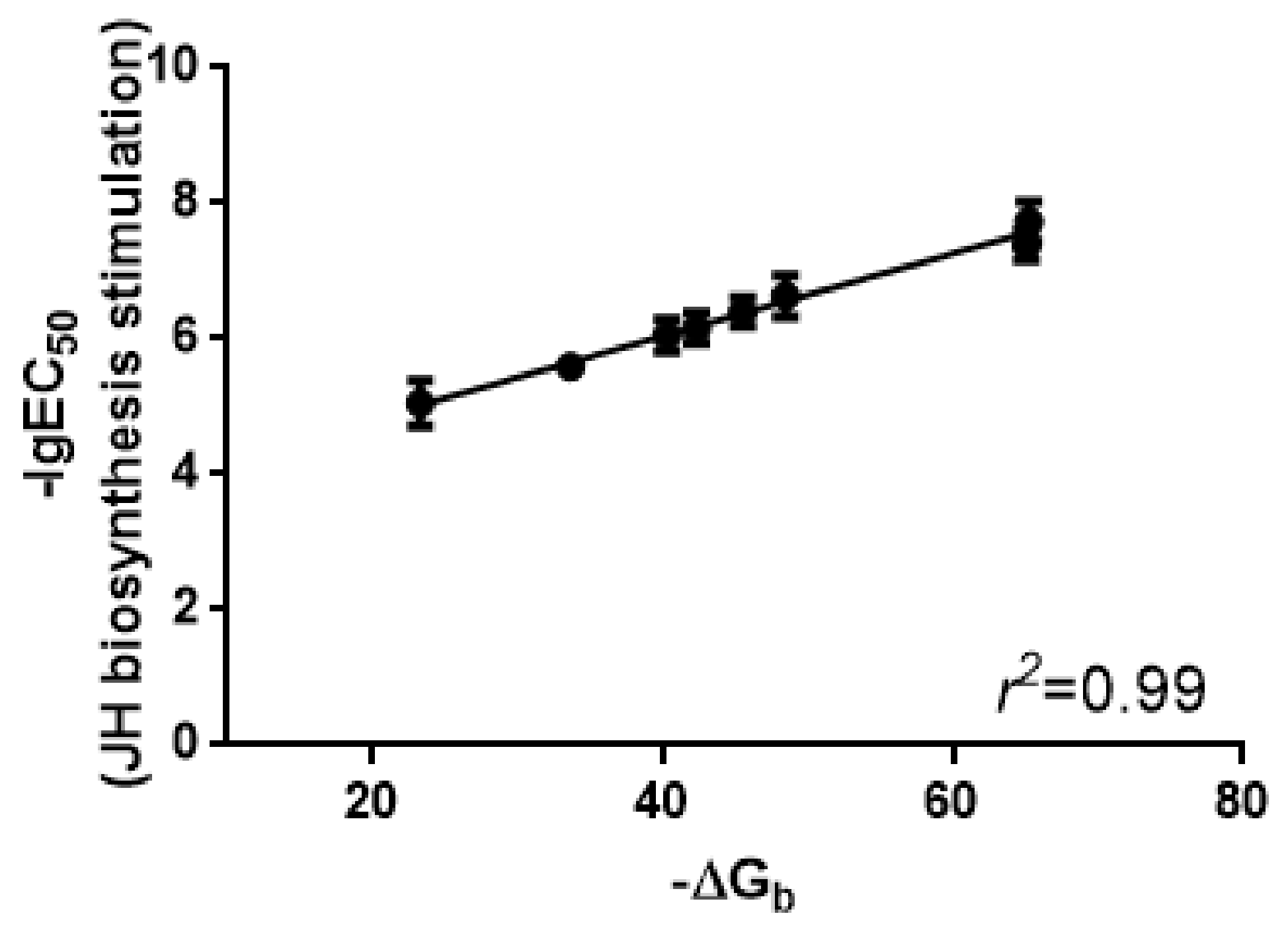
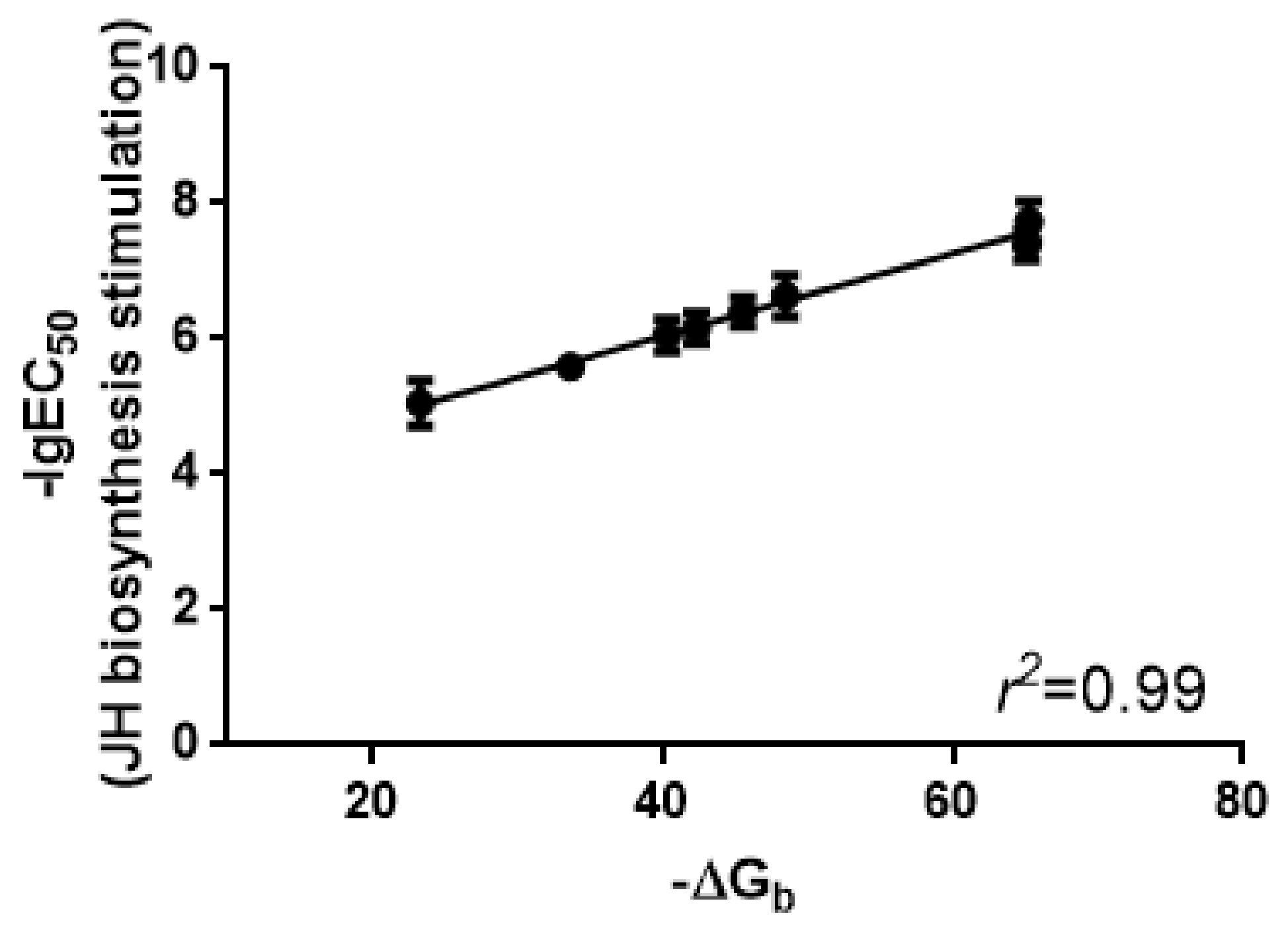
2.4. Binding Affinities of Manse-AT Analogs
2.5. Antagonist Design
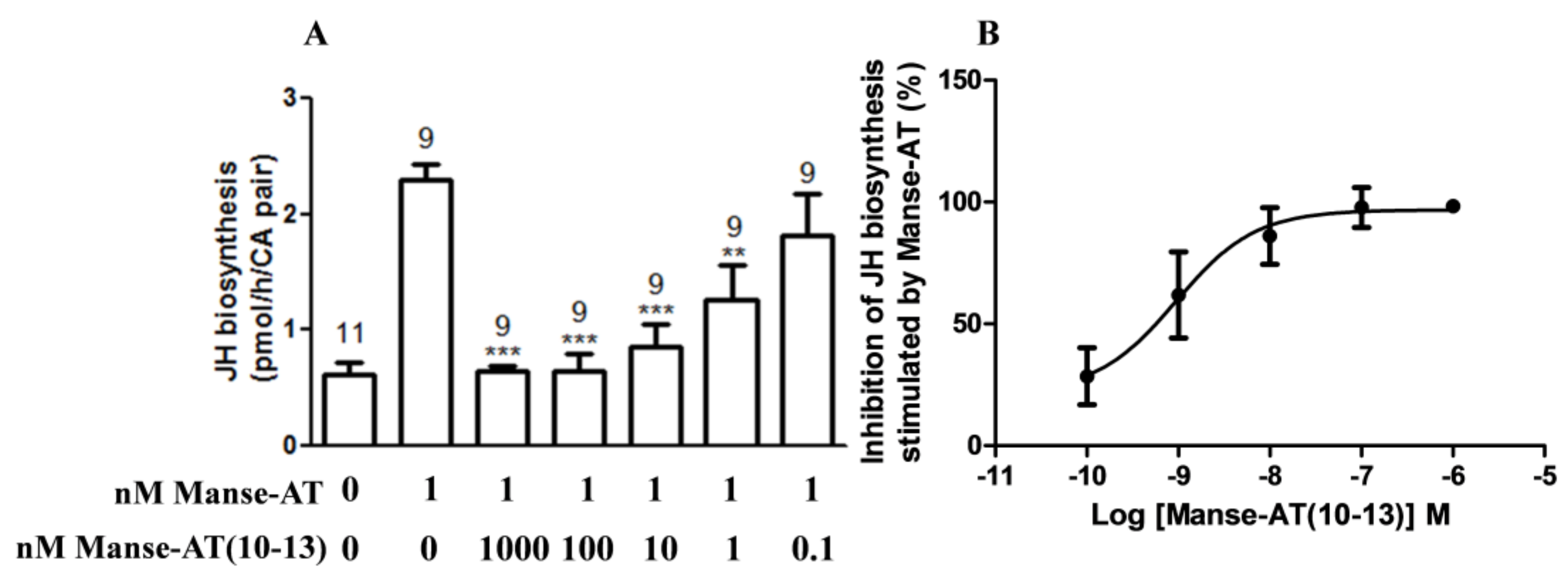
2.6. Antagonistic Effects on JH Biosynthesis
3. Materials and Methods
3.1. Chemicals
3.2. Insect
3.3. Assays for JH Biosynthesis Assays In Vitro
3.4. Peptide Synthesis
3.5. Homology Search and Modeling
3.6. Docking Calculations
3.7. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gäde, G.; Goldsworthy, G.J. Insect peptide hormones: A selective review of their physiology and potential application for pest control. Pest Manag. Sci. 2003, 59, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Nachman, R.J.; Moyna, G.; Williams, H.J.; Tobe, S.S.; Scott, A.I. Synthesis, biological activity, and conformational studies of insect allatostatin neuropeptide analogues incorporating turn-promoting moieties. Bioorg. Med. Chem. 1998, 6, 1379–1388. [Google Scholar] [CrossRef]
- Nachman, R.J.; Teal, P.E.; Aziz, O.B.; Zubrzak, P.; Altstein, M. An amphiphilic, PK/PBAN analog is a selective pheromonotropic antagonist that penetrates the cuticle of a heliothine insect. Peptides 2009, 30, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H.; Toschi, A.; Li, J.P.; Carney, R.L.; Schooley, D.A.; Kramer, S.J. Identification of an allatotropin from adult Manduca sexta. Science 1989, 243, 1481–1483. [Google Scholar] [CrossRef] [PubMed]
- Elekonich, M.E.; Horodyski, F.M. Insect allatotropins belongs to a family of structurally-related myoactive peptides present in several invertebrate phyla. Peptides 2003, 24, 1623–1632. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Horodyski, F.M.; Chamberlin, M.E. Inhibition of midgut ion transport by allatotropin (Mas-AT) and Manduca FLRFamide peptides in the tobacco hornworm, Manduca sexta. J. Exp. Biol. 1998, 201, 3067–3074. [Google Scholar] [PubMed]
- Veenstra, J.A.; Lehman, H.K.; Davis, N.T. Allatotropin is a cardioacceleratory peptide in Manduca sexta. J. Exp. Biol. 1994, 188, 347–354. [Google Scholar] [PubMed]
- Deng, X.; Kai, Z.; Chamberlin, M.E.; Horodyski, F.M.; Yang, X. The discovery of a novel antagonist—Manduca sexta allatotropin analogue—As an insect midgut active ion transport inhibitor. Pest Manag. Sci. 2016, 72, 2176–2180. [Google Scholar] [CrossRef] [PubMed]
- Horodyski, F.M.; Verlinden, H.; Filkin, N.; Vandersmissen, H.P.; Fleury, C.; Reynolds, S.E.; Kai, Z.; Broeck, J.V. Isolation and functional characterization of an allatotropin receptor from Manduca sexta. Insect Biochem. Mol. Biol. 2011, 41, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Nouzova, M.; Brockhoff, A.; Mayoral, J.G.; Goodwin, M.; Meyerhof, W.; Noriega, F.G. Functional characterization of an allatotropin receptor expressed in the corpora allata of mosquitoes. Peptides 2012, 34, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Vuerinckx, K.; Verlinden, H.; Lindemans, M.; Broeck, J.V.; Huybrechts, R. Characterization of an allatotropin-like peptide receptor in the red flour beetle Tribolium castaneum. Insect Biochem. Mol. Biol. 2011, 41, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Kobilka, B.; Schertler, G.F.X. New G-protein-coupled receptor crystal structures: Insights and limitations. Trends Pharmacol. Sci. 2008, 29, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.A.; Liu, W.; Chun, E.; Katritch, V.; Wu, H.; Vardy, E.; Huang, X.; Trapella, C.; Guerrini, R.; Calo, G.; et al. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 2012, 485, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.L.; Macarthur, M.W.; Hutchinson, E.G.; Thornton, J.M. Stereochemical quality of protein structure coordinates. Proteins 1992, 12, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.A.; Joachim, F.A. Techniques for rearing laboratory colonies of tobacco hornworms and pink bollworms. Ann. Entomol. Soc. Am. 1976, 69, 365–373. [Google Scholar] [CrossRef]
- Lee, K.Y.; Chamberlin, M.E.; Horodyski, F.M. Biological activity of Manduca sexta allatotropin-like peptides, predicted products of tissue-specific and developmentally-regulated alternatively spliced mRNAs. Peptides 2002, 23, 1933–1941. [Google Scholar] [CrossRef]
- Baker, F.C.; Tsai, L.W.; Reuter, C.C.; Schooley, D.A. In vivo, fluctuation of JH, JH acid, and ecdysteroid titer, and JH esterase activity, during development of fifth stadium Manduca sexta. Insect Biochem. 1987, 17, 989–996. [Google Scholar] [CrossRef]
- Kai, Z.; Yin, Y.; Zhang, Z.; Huang, J.; Tobe, S.S.; Chen, S. A rapid quantitative assay for juvenile hormones and intermediates in the biosynthetic pathway using gas chromatography tandem mass spectrometry. J. Chromatogr. A 2018, 1538, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.; Li, Y.; Yin, Y.; Chen, S.; Kai, Z. Discovery and quantitative structure-activity relationship study of lepidopteran HMG-CoA reductase inhibitors as selective insecticides. Pest Manag. Sci. 2017, 73, 1944–1952. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. A basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kai, Z.; Ling, Y.; Liu, W.; Zhao, F.; Yang, X. The study of solution conformation of allatostatins by 2-D NMR and molecular modeling. BBA-Proteins Proteom. 2006, 1764, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Macarthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Spitzer, R.; Jain, A.N. Surflex-Dock: Docking benchmarks and real-world application. J. Comput. Aided Mol. Des. 2012, 26, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Heller, H.; Schaefer, M.; Schulten, K. Molecular dynamics simulation of a bilayer of 200 lipids in the gel and in the liquid crystal phase. J. Phys. Chem. 1993, 97, 8343–8360. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Meyer, J.M.; Ejendal, K.F.K.; Avramova, L.; Garland-Kuntz, E.; Giraldo-Calderón, G.; Watts, V.J.; Hill, C.A. A “genome-to-lead” approach for insecticide discovery: Pharmacological characterization and chemical compound screening of Aedes aegypti D1-like dopamine receptors. PLoS Negl. Trop. Dis. 2012, 6, e1478. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | Stimulation of JH Biosynthesis | ∆Gb (kJ/mol) | |
|---|---|---|---|---|
| EC50 (nM) a (95% CI b) | Stimulation at 1 μM (Repeat Number) | |||
| Manse-AT | GFKNVEMMTARGFa | 0.9 (0.2–4.0) | 426.6 ± 86.3 (9) | |
| Manse-AT (6–13) | EMMTARGFa | 18.4 (4.8–70.5) | 600.5 ± 98.5 (9) | −65.23 |
| Manse-AT (7–13) | MMTARGFa | 228.4 (53.0–985.0) | 232.9 ± 33.7 (9) | −48.48 |
| Manse-AT (8–13) | MTARGFa | 2492 (1238–5017) | 133.1 ± 22.3 (9) | −33.63 |
| Manse-AT (9–13) | TARGFa | No effect | 10.6 ± 21.0 (11) | −7.27 |
| Manse-AT (10–13) | ARGFa | No effect | 8.1± 17.6 (13) | 3.23 c |
| Manse-AT (11–13) | RGFa | No effect | 8.3 ± 16.4 (9) | 11.03 |
| Manse-AT (6–13) [Ala1] | AMMTARGFa | 393.7 (146.9–1055) | 138.0 ± 12.7 (9) | −45.52 |
| Manse-AT (6–13) [Ala2] | EAMTARGFa | 691.6 (238.1–2009) | 89.6 ± 14.6 (9) | −42.35 |
| Manse-AT (6–13) [Ala3] | EMATARGFa | 884.3 (288.0–2715) | 91.0 ± 15.9 (9) | −40.32 |
| Manse-AT (6–13) [Ala4] | EMMAARGFa | 8958 (1803–44,500) | 10.8 ± 6.5 (9) | −23.29 |
| Manse-AT (6–13) [Ala6] | EMMTAAGFa | No effect | 10.3 ± 7.8 (9) | 10.76 |
| Manse-AT (6–13) [Ala7] | EMMTARAFa | 40.5 (13.9–118.7) | 378.4 ± 49.8 (9) | −65.09 |
| Manse-AT (6–13) [Ala8] | EMMTARGAa | No effect | 4.5 ± 8.5 (9) | 7.54 |
| No. | Structure | Antagonistic Effects on JH Biosynthesis IC50 (nM) a (95% CI b) |
|---|---|---|
| Manse-AT (10–13) | Ala-Arg-Gly-Phe-NH2 | 0.9 (0.2–5.4) |
| 1 | Gly-Arg-Gly-Phe-NH2 | 1.2 (0.4–3.8) |
| 2 | Leu-Arg-Gly-Phe-NH2 | 35.4 (15.9–78.6) |
| 3 | 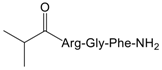 | 26.1 (8.8–78.1) |
| 4 | 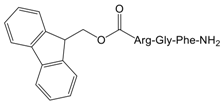 | No effect |
| 5 | Ala-Ala-Gly-Phe-NH2 | No effect |
| 6 | Ala-Leu-Gly-Phe-NH2 | No effect |
| 7 | Ala-Arg-Ala-Phe-NH2 | 17.5 (8.3–37.1) |
| 8 | 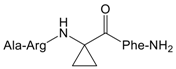 | 1031 (305.5–3479) |
| 9 | Ala-Arg-Gly-Ala-NH2 | No effect |
| 10 | Ala-Arg-Gly-Tyr-NH2 | 1.0 (0.4–2.5) |
| 11 | 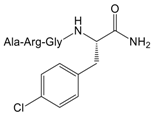 | 8.4 (3.9–18.3) |
| 12 | 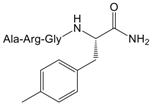 | 9.4 (3.2–27.1) |
| Manse-AT (11–13) | Arg-Gly-Phe-NH2 | No effect |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kai, Z.-P.; Zhu, J.-J.; Deng, X.-L.; Yang, X.-L.; Chen, S.-S. Discovery of a Manduca sexta Allatotropin Antagonist from a Manduca sexta Allatotropin Receptor Homology Model. Molecules 2018, 23, 817. https://doi.org/10.3390/molecules23040817
Kai Z-P, Zhu J-J, Deng X-L, Yang X-L, Chen S-S. Discovery of a Manduca sexta Allatotropin Antagonist from a Manduca sexta Allatotropin Receptor Homology Model. Molecules. 2018; 23(4):817. https://doi.org/10.3390/molecules23040817
Chicago/Turabian StyleKai, Zhen-Peng, Jing-Jing Zhu, Xi-Le Deng, Xin-Ling Yang, and Shan-Shan Chen. 2018. "Discovery of a Manduca sexta Allatotropin Antagonist from a Manduca sexta Allatotropin Receptor Homology Model" Molecules 23, no. 4: 817. https://doi.org/10.3390/molecules23040817
APA StyleKai, Z.-P., Zhu, J.-J., Deng, X.-L., Yang, X.-L., & Chen, S.-S. (2018). Discovery of a Manduca sexta Allatotropin Antagonist from a Manduca sexta Allatotropin Receptor Homology Model. Molecules, 23(4), 817. https://doi.org/10.3390/molecules23040817