Computational Simulation Studies on the Binding Selectivity of 1-(1H-Benzimidazol-5-yl)-5-aminopyrazoles in Complexes with FGFR1 and FGFR4

Abstract
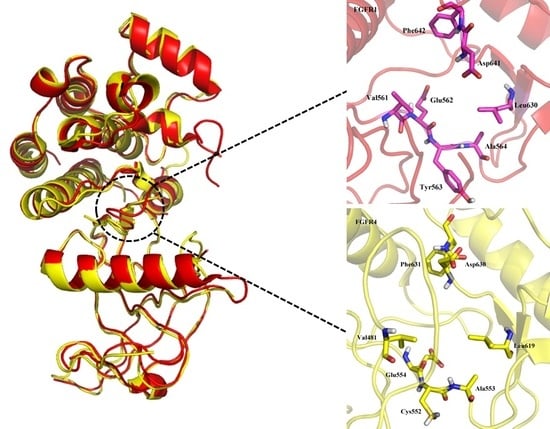
:
1. Introduction
2. Computational Details
2.1. Preparation of Initial Complex
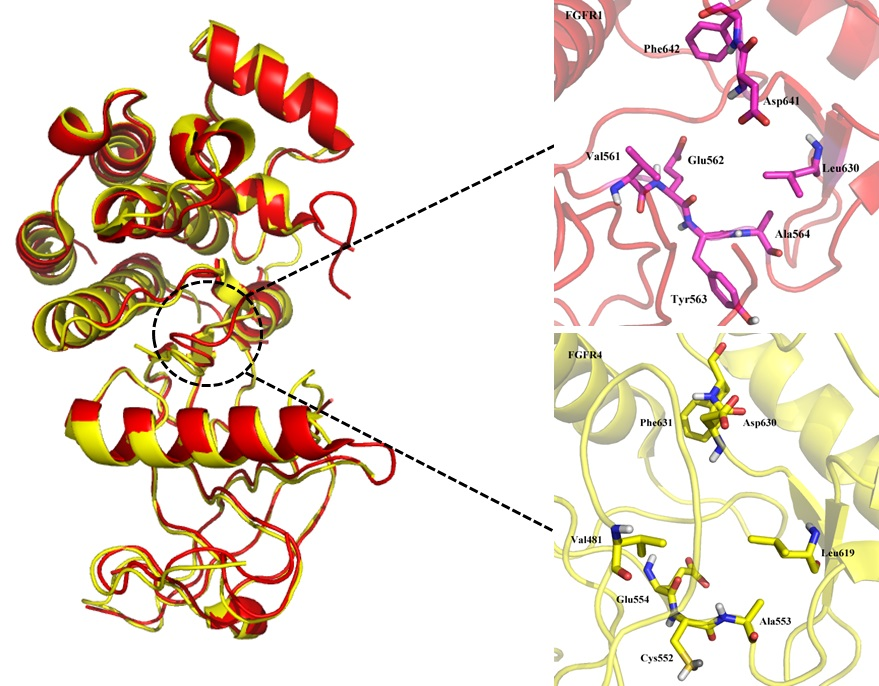
2.1.1. Binding Site Comparison
2.1.2. Docking Simulations
2.1.3. Molecular Dynamics Simulations
2.1.4. Binding Free Energy Calculations and Per-Residue Free Energy Decomposition Analyses
2.1.5. Molecular Electrostatic Potential (MESP) Analyses
3. Results and Discussion
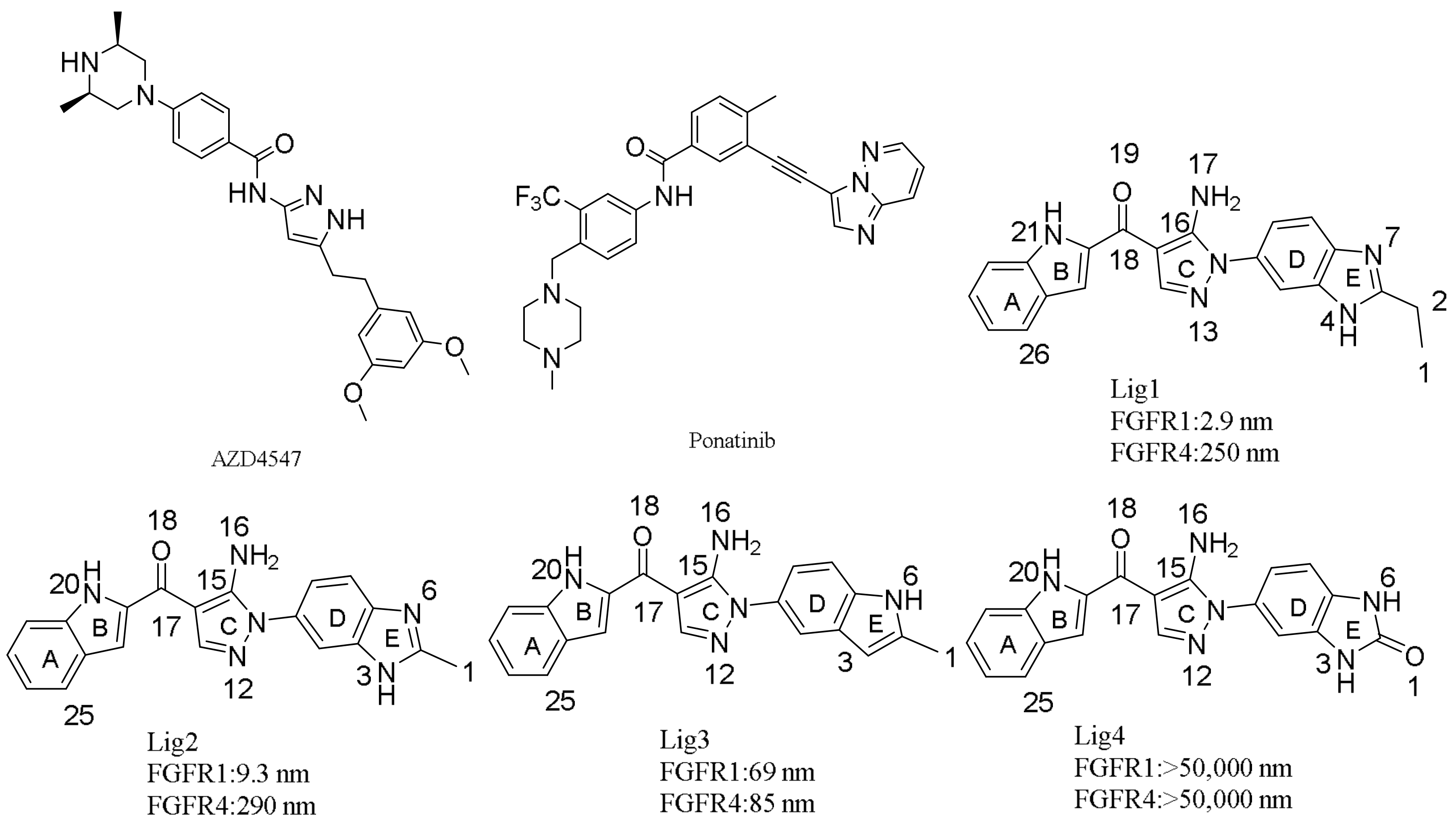
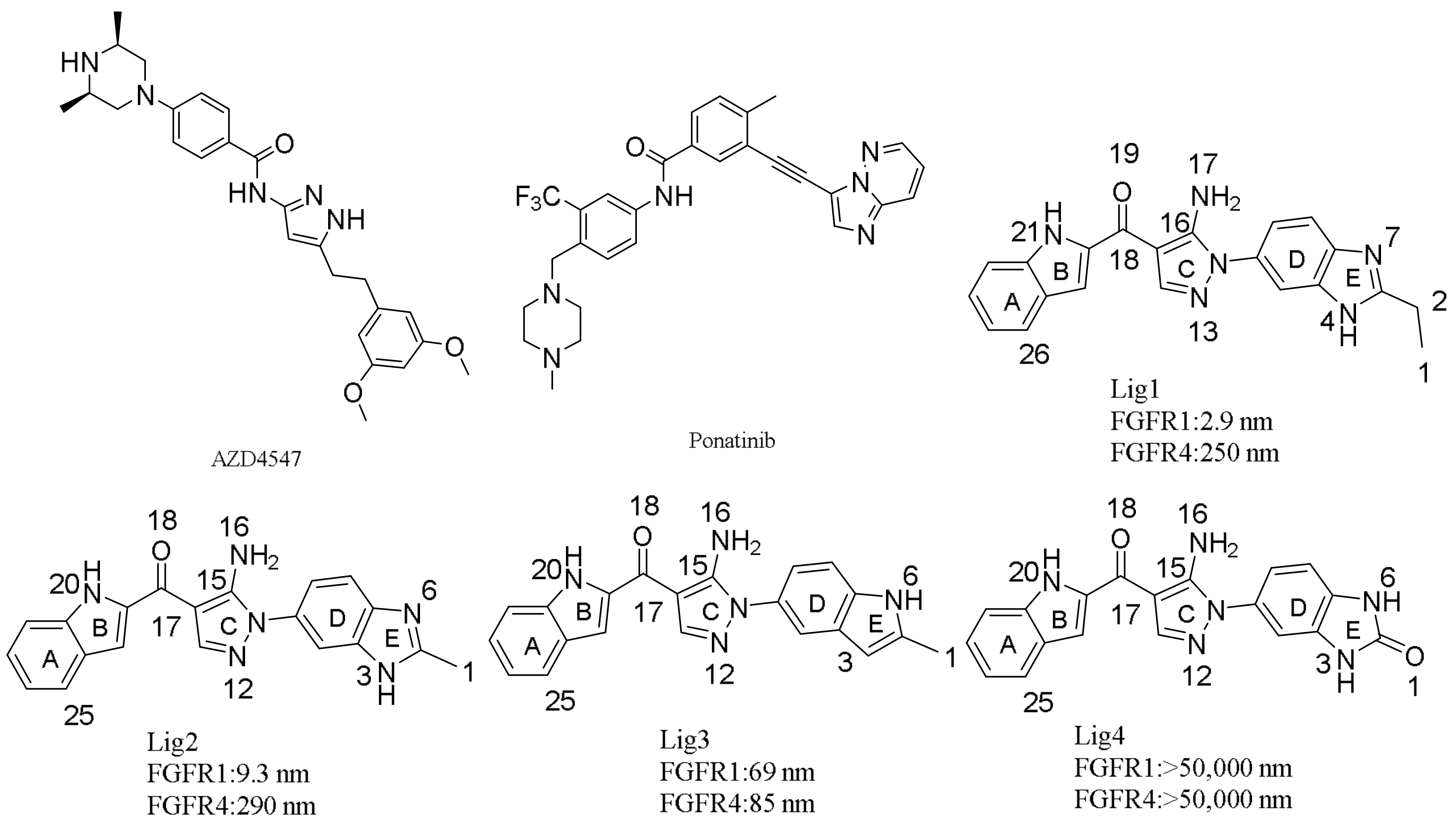
3.1. Molecular Docking
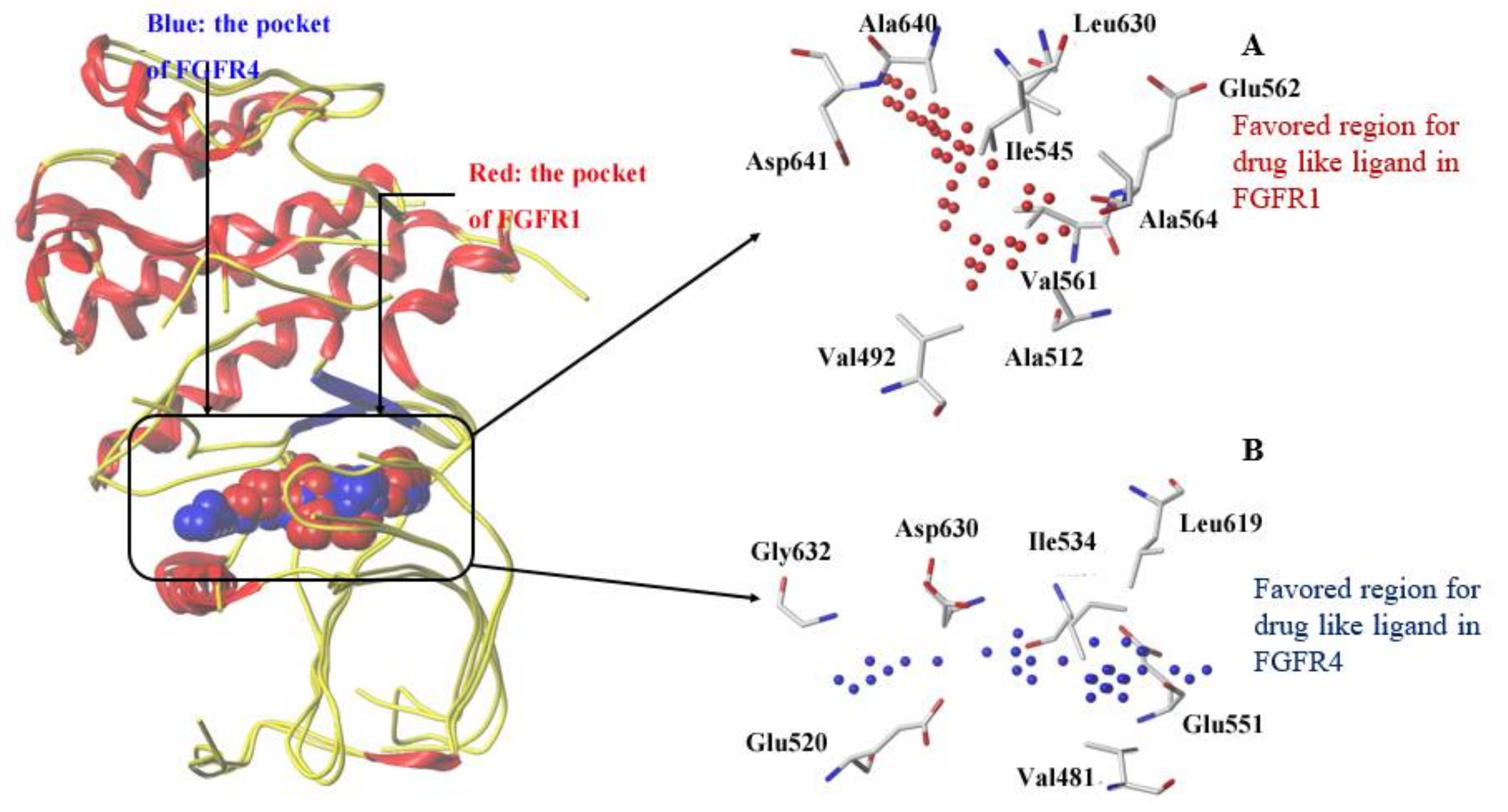
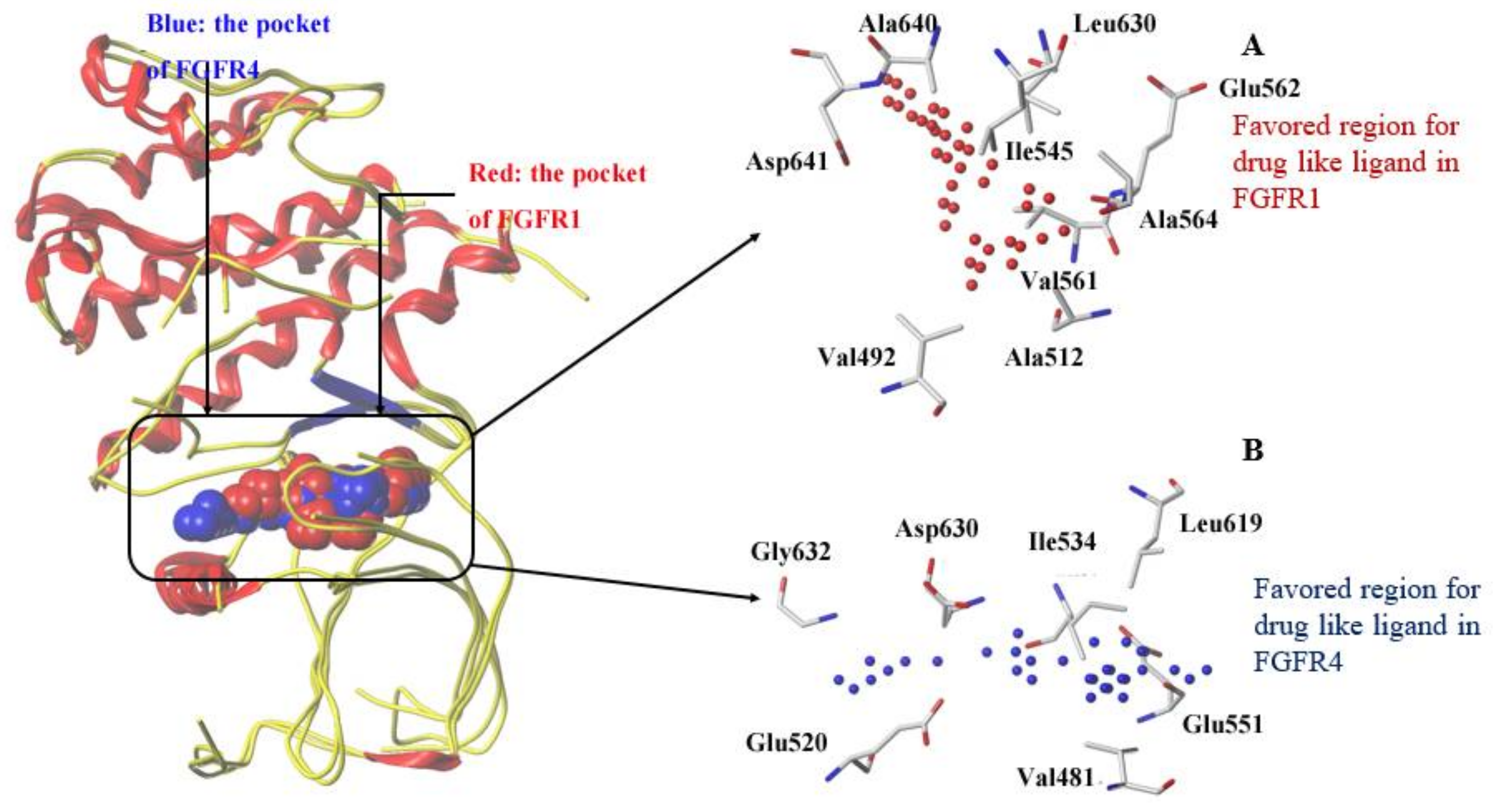
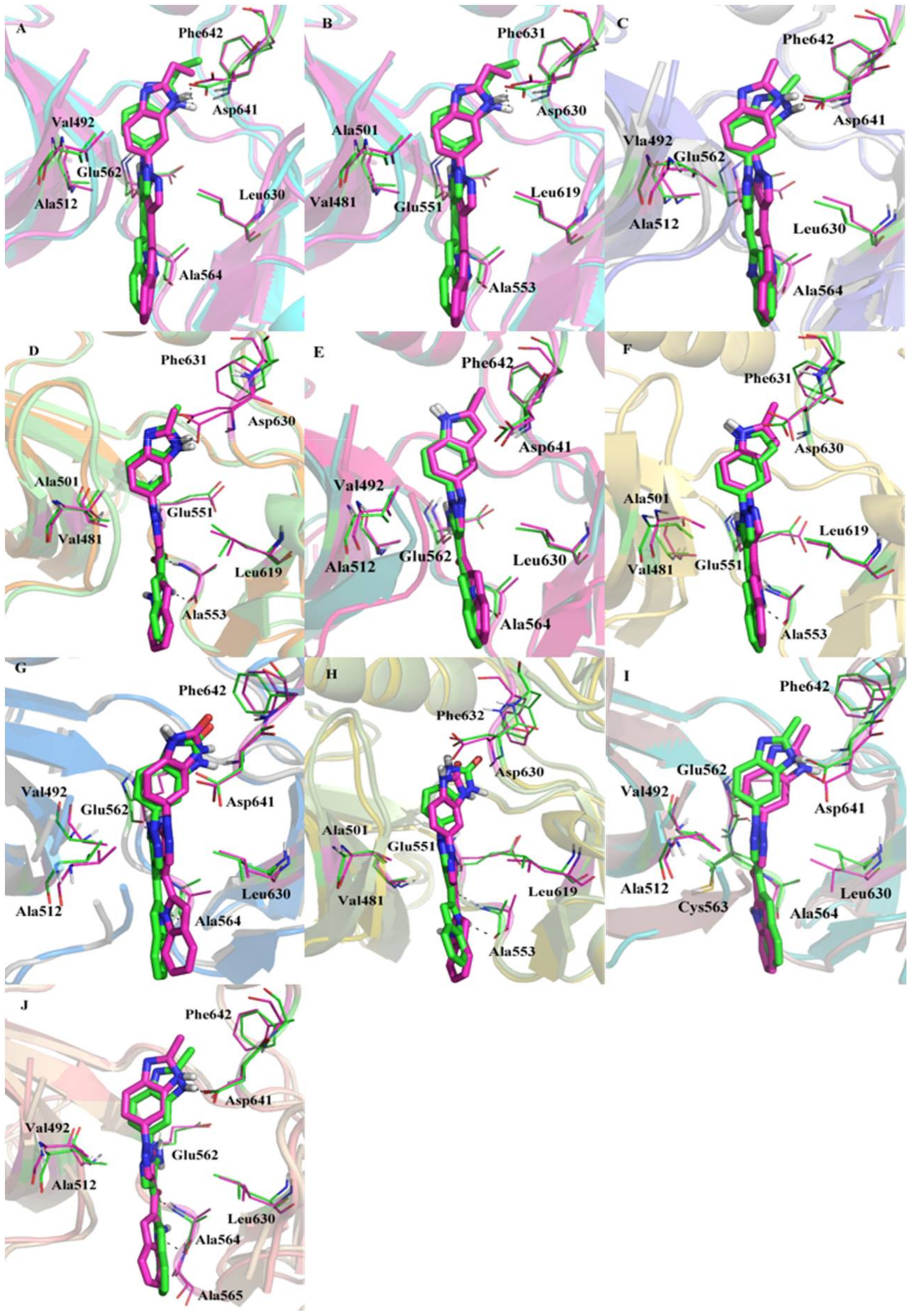
3.2. Comparison of Binding Sites of FGFR1 and FGFR4
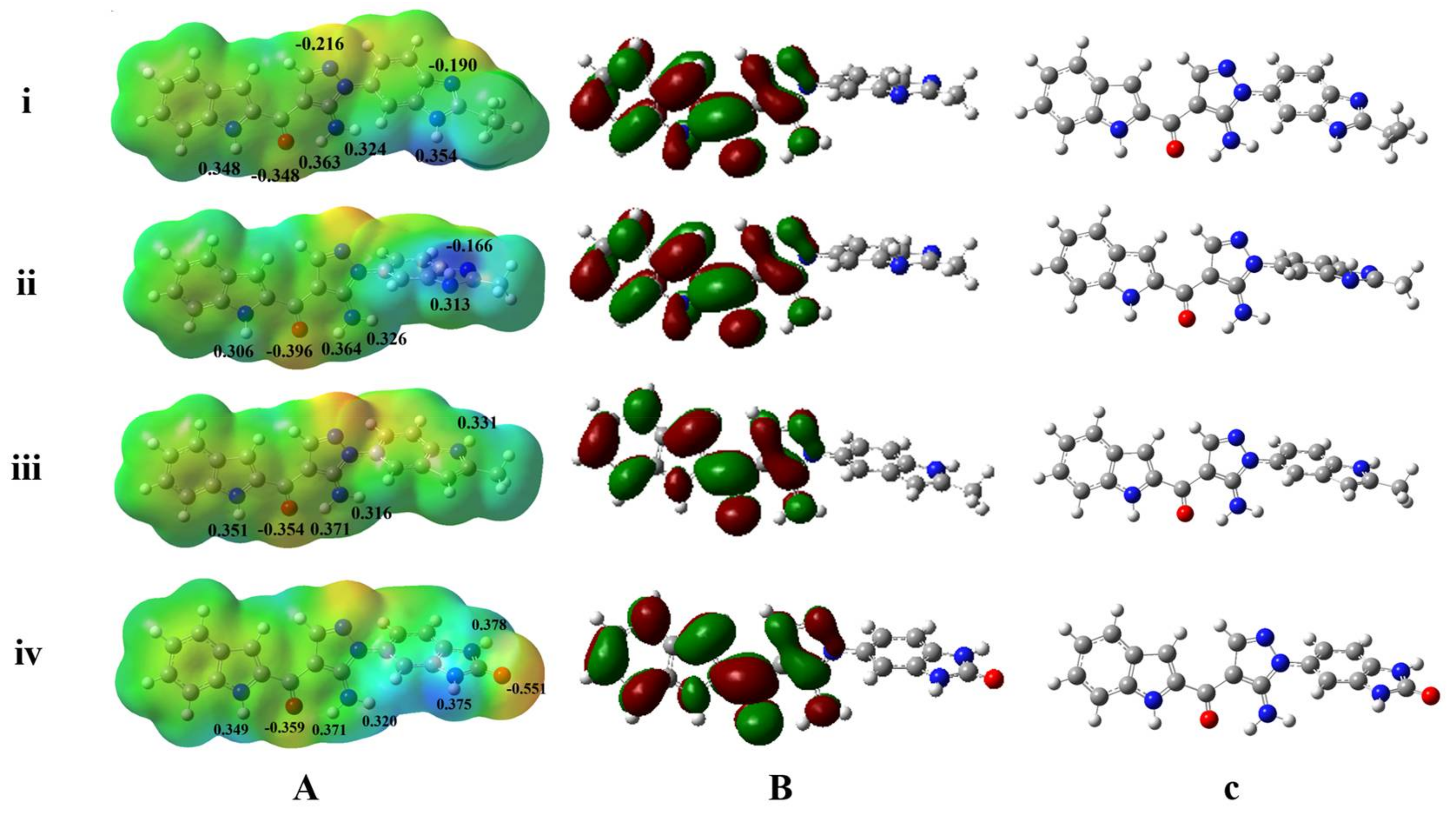
3.3. MESP Potential Surface Analyses
3.4. HOMO and LUMO Orbitals Analyses
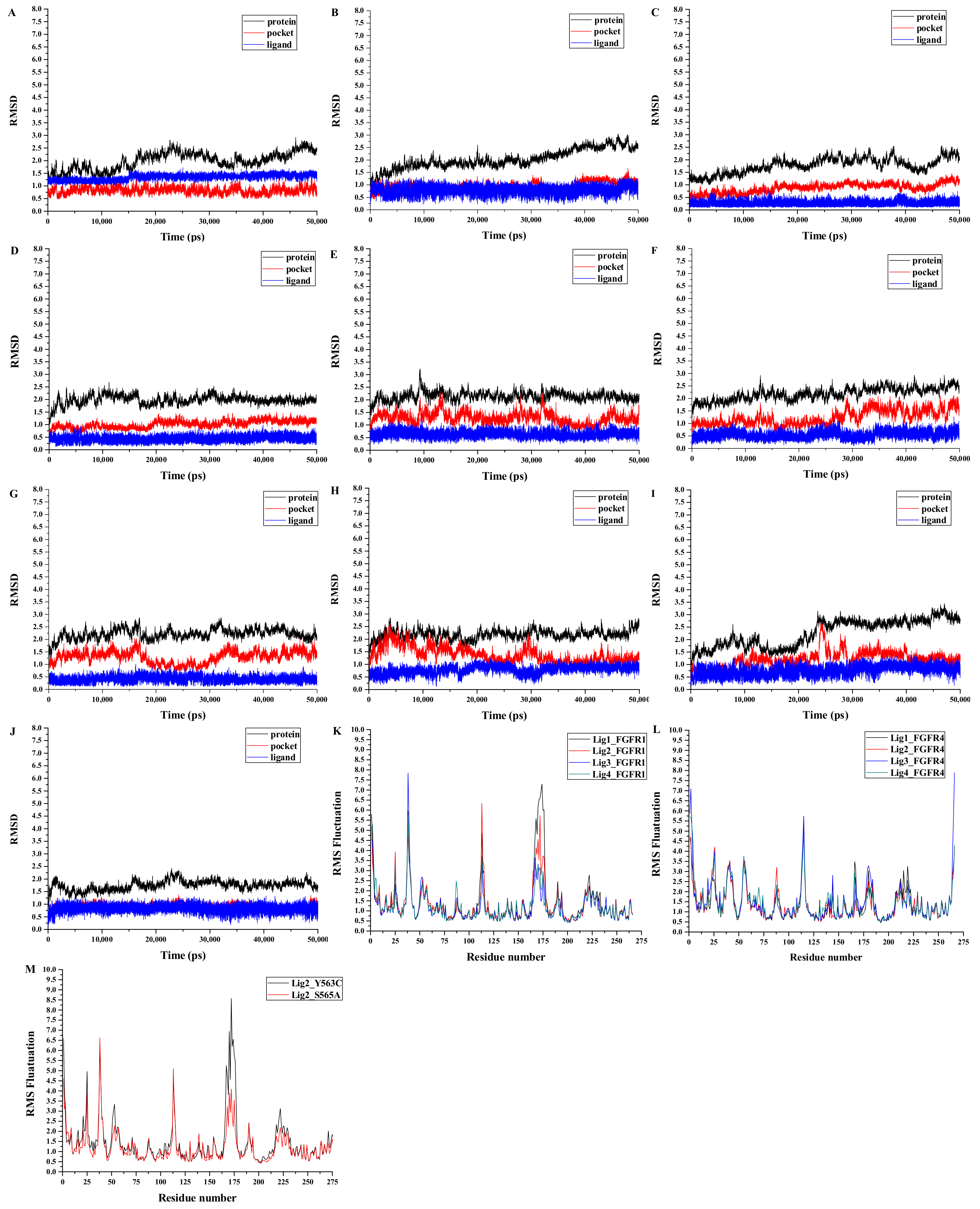
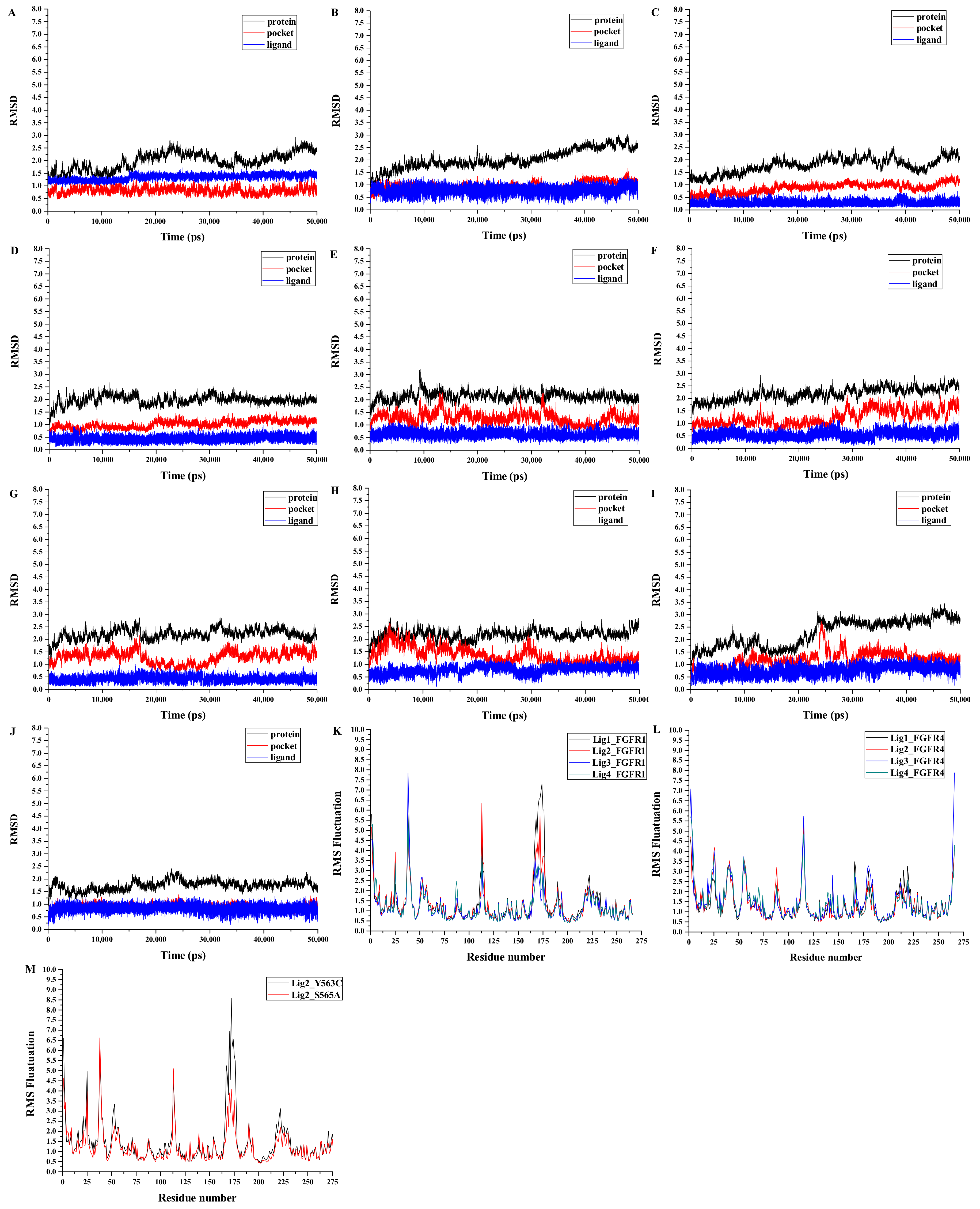
3.5. Analyses of Structure Stability and Flexibility
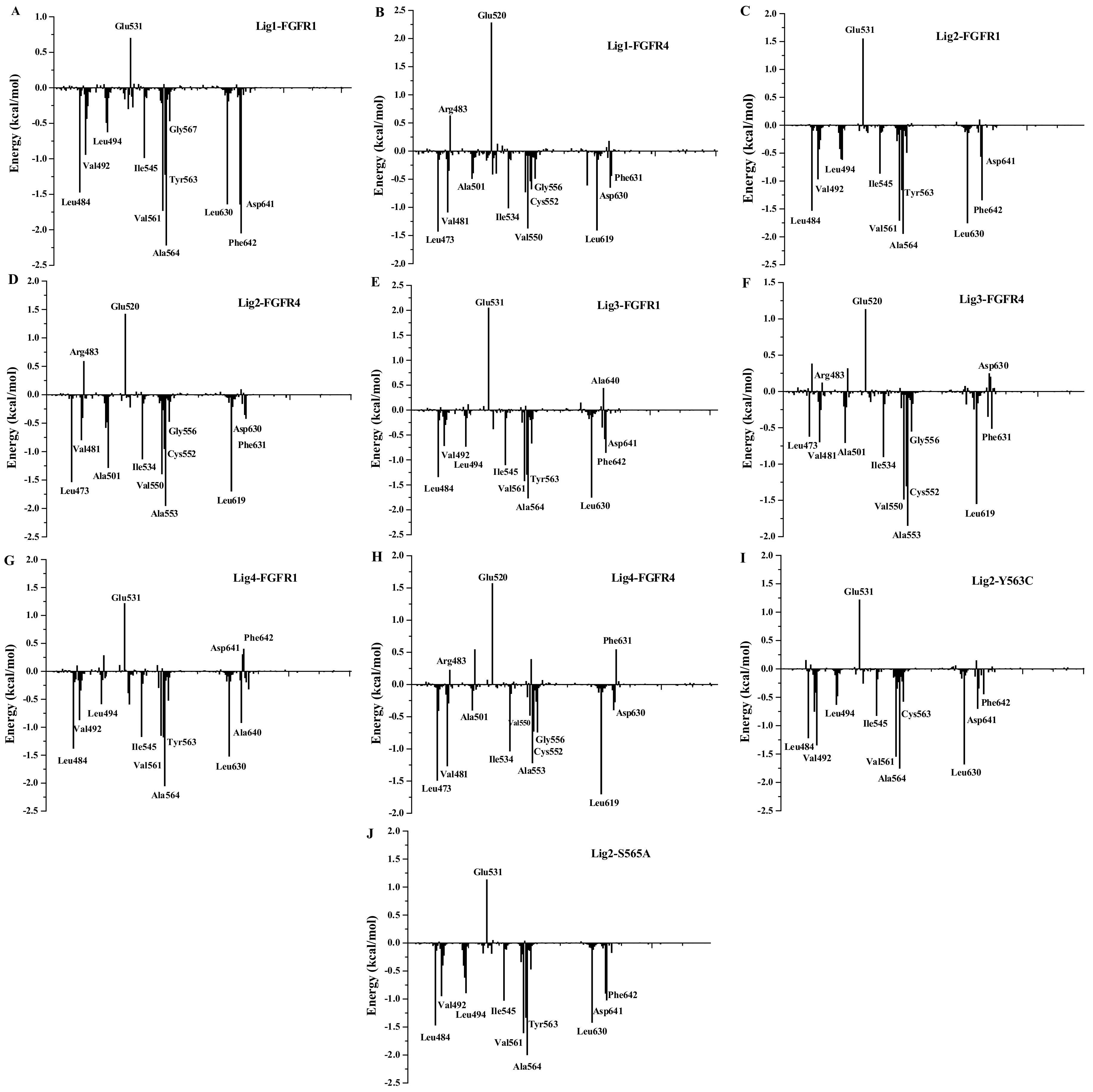
3.6. Binding Free Energy Calculations
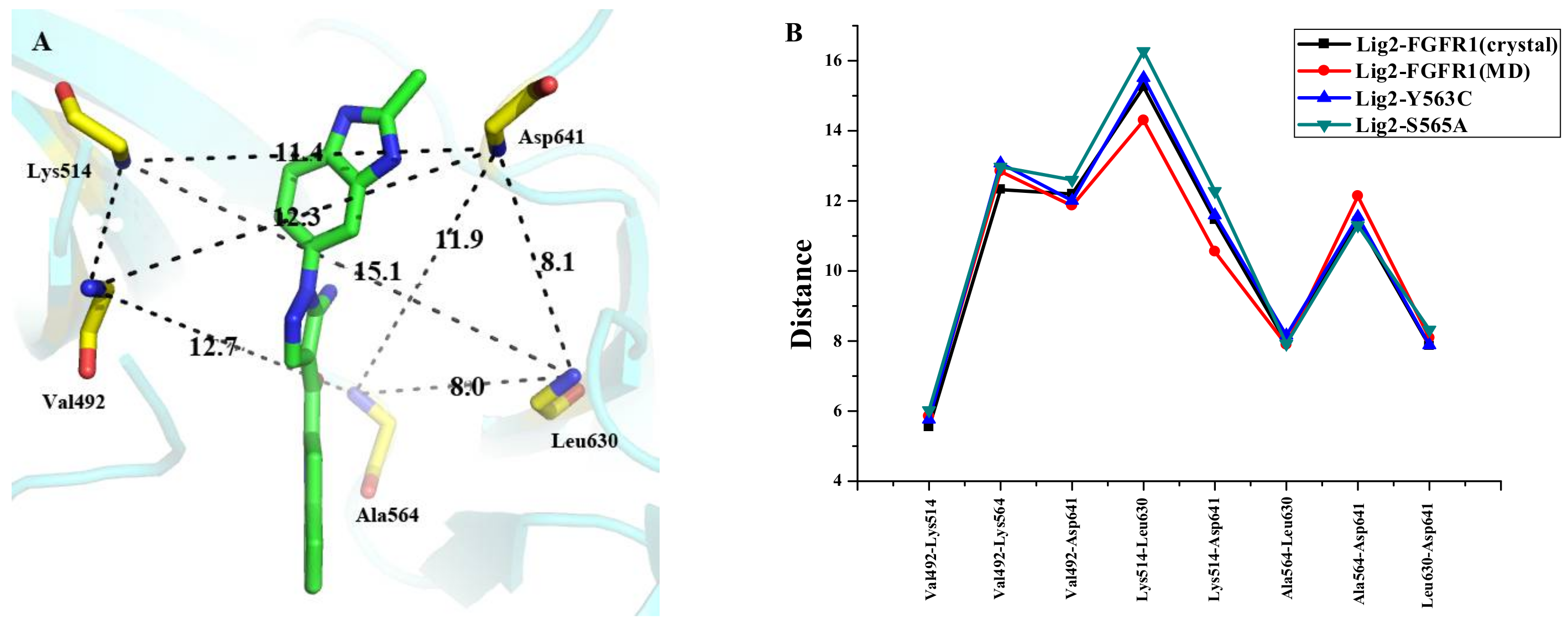
3.7. Dynamic Analyses of the Selectivity Mechanism of Inhibitors
3.8. Insight to the Activity Difference of Lig1 and Lig4 to FGFR1
3.9. Insight to the Activity and Selectivity of Lig1 in Complexes Lig1-FGFR1 and Lig1-FGFR4
3.10. Insight to the Activity and Selectivity of Lig2 in Complexes Lig2-FGFR1, Lig2-Mutants, and Lig2-FGFR4
3.11. Insight to the Bioactivity and Selectivity of Lig1 and Lig3 Binding to FGFR1 in Complexes of Lig1-FGFR1 and Lig3-FGFR1
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Hallinan, N.; Finn, S.; Cuffe, S.; Rafee, S.; O’Byrne, K.; Gately, K. Targeting the fibroblast growth factor receptor family in cancer. Cancer Treat. Rev. 2016, 46, 51–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touat, M.; Ileana, E.; Postelvinay, S.; André, F.; Soria, J.C. Targeting FGFR Signaling in Cancer. Clin. Cancer Res. 2015, 21, 2684–2694. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; Lecouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- Guagnano, V.; Furet, P.; Spanka, C.; Bordas, V.; Le Douget, M.; Stamm, C.; Brueggen, J.; Jensen, M.R.; Schnell, C.; Schmid, H. Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J. Med. Chem. 2011, 54, 7066–7083. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Wang, M.; Tian, X.; Zhang, X. An overview of the binding models of FGFR tyrosine kinases in complex with small molecule inhibitors. Eur. J. Med. Chem. 2016, 126, 476. [Google Scholar] [CrossRef] [PubMed]
- André, F.; Cortés, J. Rationale for targeting fibroblast growth factor receptor signaling in breast cancer. Breast Cancer Res. Treat. 2015, 150, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Adjei, A.A. FGFR Signaling as a Target for Lung Cancer Therapy. J. Thorac. Oncol. 2016, 11, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Su, X.; Zhang, L.; Yin, X.; Tang, L.; Zhang, X.; Xu, Y.; Gao, Z.; Liu, K.; Zhou, M. FGFR2 gene amplification in gastric cancer predicts sensitivity to the selective FGFR inhibitor AZD4547. Clin. Cancer Res. 2013, 19, 2572–2583. [Google Scholar] [CrossRef] [PubMed]
- Manetti, F.; Botta, M. Small-molecule inhibitors of fibroblast growth factor receptor (FGFR) tyrosine kinases (TK). Curr. Pharm. Des. 2003, 9, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Liu, Z.; Wu, J.; Cai, Y.; Li, X. Anticancer molecules targeting fibroblast growth factor receptors. Trends Pharmacol. Sci. 2012, 33, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Tiseo, M.; Gelsomino, F.; Alfieri, R.; Cavazzoni, A.; Bozzetti, C.; De Giorgi, A.M.; Petronini, P.G.; Ardizzoni, A. FGFR as potential target in the treatment of squamous non small cell lung cancer. Cancer Treat. Rev. 2015, 41, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J.; Plotnikov, A.N.; Ibrahimi, O.A.; Eliseenkova, A.V.; Yeh, B.K.; Yayon, A.; Linhardt, R.J.; Mohammadi, M. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell 2000, 6, 743–750. [Google Scholar] [CrossRef]
- Lesca, E.; Lammens, A.; Huber, R.; Augustin, M. Structural analysis of the human fibroblast growth factor receptor 4 kinase. J. Mol. Biol. 2014, 426, 3744–3756. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Tan, L.; Wang, H.; Liu, Y.; Blais, S.; Deng, J.; Neubert, T.A.; Gray, N.S.; Li, X.; Mohammadi, M. DFG-out mode of inhibition by an irreversible type-1 inhibitor capable of overcoming gate-keeper mutations in FGF receptors. ACS Chem. Biol. 2014, 10, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Chen, L.; Wang, Z.; Kang, Y.; Wu, C.; Xia, Q.; Liu, Z.; Zhou, J.; Liang, G.; Cai, Y. Theoretical studies on FGFR isoform selectivity of FGFR1/FGFR4 inhibitors by molecular dynamics simulations and free energy calculations. Phys. Chem. Chem. Phys. 2017, 19, 3649–3659. [Google Scholar] [CrossRef] [PubMed]
- Tucker, J.A.; Klein, T.; Breed, J.; Breeze, A.L.; Overman, R.; Phillips, C.; Norman, R.A. Structural insights into FGFR kinase isoform selectivity: Diverse binding modes of AZD4547 and ponatinib in complex with FGFR1 and FGFR4. Structure 2014, 22, 1764–1774. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis. Int. J. Mol. Med. 2016, 38, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Commodore, L.; Huang, W.S.; Wang, Y.; Thomas, M.; Keats, J.; Xu, Q.; Rivera, V.M.; Shakespeare, W.C.; Clackson, T. Structural Mechanism of the Pan-BCR-ABL Inhibitor Ponatinib (AP24534): Lessons for Overcoming Kinase Inhibitor Resistance. Chem. Biol. Drug Des. 2011, 77, 1–11. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, T.; Shakespeare, W.C.; Zhu, X.; Eide, C.A.; Rivera, V.M.; Wang, F.; Adrian, L.T.; Zhou, T.; Huang, W.S.; Xu, Q. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 2009, 16, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Gavine, P.R.; Mooney, L.; Kilgour, E.; Thomas, A.P.; Al-Kadhimi, K.; Beck, S.; Rooney, C.; Coleman, T.; Baker, D.; Mellor, M.J. AZD4547: An orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res. 2012, 72, 2045–2056. [Google Scholar] [CrossRef] [PubMed]
- Mo, C.; Zhang, Z.; Guise, C.P.; Li, X.; Luo, J.; Tu, Z.; Xu, Y.; Patterson, A.V.; Smaill, J.B.; Ren, X. 2-Aminopyrimidine derivatives as new selective fibroblast growth factor receptor 4 (FGFR4) inhibitors. ACS Med. Chem. Lett. 2017, 8, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Norman, R.A.; Schott, A.K.; Andrews, D.M.; Breed, J.; Foote, K.M.; Garner, A.P.; Ogg, D.; Orme, J.P.; Pink, J.H.; Roberts, K. Protein-ligand crystal structures can guide the design of selective inhibitors of the FGFR tyrosine kinase. J. Med. Chem. 2012, 55, 5003. [Google Scholar] [CrossRef] [PubMed]
- Posy, S.L.; Hermsmeier, M.A.; Vaccaro, W.; Ott, K.H.; Todderud, G.; Lippy, J.S.; Trainor, G.L.; Loughney, D.A.; Johnson, S.R. Trends in kinase selectivity: Insights for target class-focused library screening. J. Med. Chem. 2011, 54, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Ebiike, H.; Taka, N.; Matsushita, M.; Ohmori, M.; Takami, K.; Hyohdoh, I.; Kohchi, M.; Hayase, T.; Nishii, H.; Morikami, K. Discovery of [5-Amino-1-(2-methyl-3H-benzimidazol-5-yl) pyrazol-4-yl]-(1H-indol-2-yl) methanone (CH5183284/Debio 1347), An Orally Available and Selective Fibroblast Growth Factor Receptor (FGFR) Inhibitor. J. Med. Chem. 2016, 59, 10586–10600. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.N. Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Cruciani, G.; Watson, K.A. Comparative molecular field analysis using GRID force-field and GOLPE variable selection methods in a study of inhibitors of glycogen phosphorylase b. J. Med. Chem. 1994, 37, 2589–2601. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.M.; Marshall, G.R. Cavity search: An algorithm for the isolation and display of cavity-like binding regions. J. Comput.-Aided Mol. Des. 1990, 4, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Akula, N.; Lecanu, L.; Greeson, J.; Papadopoulos, V. 3D QSAR studies of AChE inhibitors based on molecular docking scores and CoMFA. Bioorg. Med. Chem. Lett. 2006, 16, 6277. [Google Scholar] [CrossRef] [PubMed]
- Case, D.; Darden, T.; Cheatham, T., III; Simmerling, C.; Wang, J.; Duke, R.; Luo, R.; Walker, R.; Zhang, W.; Merz, K. Amber Tools 13 and Amber 12; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Case, D.A.; Babin, V.; Berryman, J.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. Amber 14. Mech. Eng. 2014, 126, 14. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Computat. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Fogolari, F.; Brigo, A.; Molinari, H. Protocol for MM/PBSA molecular dynamics simulations of proteins. Biophys. J. 2003, 85, 159–166. [Google Scholar] [CrossRef]
- Chohan, T.A.; Chen, J.J.; Qian, H.Y.; Pan, Y.L.; Chen, J.Z. Molecular modeling studies to characterize N-phenylpyrimidin-2-amine selectivity for CDK2 and CDK4 through 3D-QSAR and molecular dynamics simulations. Mol. Biosyst. 2016, 12, 1250–1268. [Google Scholar] [CrossRef] [PubMed]
- Chohan, T.A.; Qian, H.Y.; Pan, Y.L.; Chen, J.Z. Molecular simulation studies on the binding selectivity of 2-anilino-4-(thiazol-5-yl)-pyrimidines in complexes with CDK2 and CDK7. Mol. Biosyst. 2016, 12, 145. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.; Trucks, G.; Schlegel, H.B.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09, Revision D. 01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Stewart, J.J. Optimization of parameters for semiempirical methods I. Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef]
- Becke, A.D. Becke’s three parameter hybrid method using the LYP correlation functional. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex System | Lig1-FGFR1 | Lig2-FGFR1 | Lig3-FGFR1 | Lig4-FGFR1 | Lig1-FGFR4 | Lig2-FGFR4 | Lig3-FGFR4 | Lig4-FGFR4 | Lig2-Y563C d | Lig2-S565A d |
|---|---|---|---|---|---|---|---|---|---|---|
| ΔEvdW | −54.86 ± 2.42 | −45.82 ± 2.53 | −44.76 ± 2.45 | −41.53 ± 2.67 | −45.81 ± 2.65 | −41.76 ± 2.77 | −42.27 ± 2.87 | −39.21 ± 2.94 | −41.96 ± 2.57 | −43.04 ± 2.75 |
| ΔEele | −15.61 ± 2.64 | −22.93 ± 2.82 | −24.96 ± 3.09 | −28.95 ± 4.67 | −24.57 ± 3.48 | −27.92 ± 5.18 | −25.32 ± 3.90 | −20.66 ± 9.64 | −17.54 ± 3.02 | −23.01 ± 2.80 |
| ΔGnonpol,sol | −4.21 ± 0.10 | −4.16 ± 0.09 | −4.10 ± 0.10 | −3.96 ± 0.09 | −4.32 ± 0.11 | −4.19 ± 0.10 | −4.29 ± 0.11 | −4.01 ± 0.13 | −4.20 ± 0.10 | −4.08 ± 0.09 |
| ΔGele,sol(PB) | 30.58 ± 3.03 | 37.40 ± 3.26 | 38.10 ± 3.12 | 38.81 ± 3.08 | 40.78 ± 4.87 | 40.80 ± 5.02 | 37.01 ± 4.59 | 31.98 ± 7.94 | 31.53 ± 3.59 | 34.16 ± 2.58 |
| ΔGele,sol(GB) | 32.23 ± 2.99 | 38.20 ± 2.99 | 36.21 ± 2.56 | 36.52 ± 2.60 | 37.41 ± 2.93 | 39.87 ± 4.39 | 31.34 ± 3.21 | 32.27 ± 7.67 | 31.31 ± 2.36 | 36.30 ± 2.52 |
| ΔEvdW + ΔGnonpol,sol | −59.07 ± 2.52 | −49.98 ± 2.62 | −48.86 ± 2.55 | −45.49 ± 2.76 | −50.13 ± 2.76 | −45.95 ± 2.87 | −46.56 ± 2.98 | −43.22 ± 3.07 | −46.16 ± 2.67 | −47.12 ± 2.84 |
| ΔEele + ΔGele,sol(PB) | 14.97 ± 5.67 | 14.47 ± 6.08 | 13.14 ± 6.21 | 9.86 ± 7.75 | 16.21 ± 6.35 | 12.88 ± 6.42 | 11.69 ± 6.31 | 11.32 ± 7.89 | 13.99 ± 6.61 | 11.15 ± 5.38 |
| ΔEele + ΔGele,sol(GB) | 16.62 ± 5.63 | 15.27 ± 5.81 | 7.25 ± 5.65 | 7.57 ± 7.27 | 12.84 ± 6.41 | 11.95 ± 5.47 | 8.02 ± 5.71 | 11.61 ± 7.51 | 13.77 ± 5.38 | 13.29 ± 5.32 |
| TΔS | −22.07 ± 6.23 | −20.01 ± 6.49 | −25.53 ± 3.63 | −28.25 ± 6.33 | −21.39 ± 4.63 | −21.86 ± 5.42 | −22.83 ± 5.67 | −26.66 ± 8.09 | −23.15 ± 7.17 | −23.86 ± 5.74 |
| ΔGpred(PB) | −22.03 ± 1.96 | −15.50 ± 2.21 | −10.19 ± 4.13 | −7.38 ± 3.18 | −12.53 ± 3.48 | −11.21 ± 3.87 | −15.71 ± 3.62 | −5.24 ± 2.87 | −9.02 ± 2.11 | −12.11 ± 2.48 |
| ΔGpred(GB) | −20.38 ± 1.92 | −14.70 ± 1.94 | −12.08 ± 3.57 | −9.67 ± 3.70 | −15.9 ± 3.54 | −12.14 ± 2.92 | −17.71 ± 3.02 | −4.95 ± 2.49 | −9.24 ± 0.88 | −9.97 ± 2.42 |
| IC50 (nM) b | 2.9 | 9.3 | 69 | >50,000 | 250 | 290 | 85 | >50,000 | NA | NA |
| ΔGexp c | −11.71 | −11.02 | −9.82 | NA | −9.06 | −8.97 | −9.70 | NA | NA | NA |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, Y.-L.; Liu, Y.-L.; Chen, J.-Z. Computational Simulation Studies on the Binding Selectivity of 1-(1H-Benzimidazol-5-yl)-5-aminopyrazoles in Complexes with FGFR1 and FGFR4. Molecules 2018, 23, 767. https://doi.org/10.3390/molecules23040767
Pan Y-L, Liu Y-L, Chen J-Z. Computational Simulation Studies on the Binding Selectivity of 1-(1H-Benzimidazol-5-yl)-5-aminopyrazoles in Complexes with FGFR1 and FGFR4. Molecules. 2018; 23(4):767. https://doi.org/10.3390/molecules23040767
Chicago/Turabian StylePan, You-Lu, Yan-Ling Liu, and Jian-Zhong Chen. 2018. "Computational Simulation Studies on the Binding Selectivity of 1-(1H-Benzimidazol-5-yl)-5-aminopyrazoles in Complexes with FGFR1 and FGFR4" Molecules 23, no. 4: 767. https://doi.org/10.3390/molecules23040767
APA StylePan, Y.-L., Liu, Y.-L., & Chen, J.-Z. (2018). Computational Simulation Studies on the Binding Selectivity of 1-(1H-Benzimidazol-5-yl)-5-aminopyrazoles in Complexes with FGFR1 and FGFR4. Molecules, 23(4), 767. https://doi.org/10.3390/molecules23040767