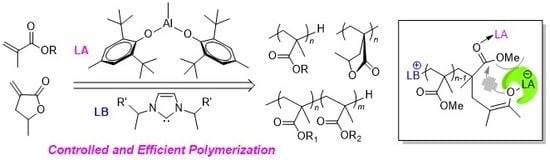
Controlled and Efficient Polymerization of Conjugated Polar Alkenes by Lewis Pairs Based on Sterically Hindered Aryloxide-Substituted Alkylaluminum

Abstract
:
1. Introduction
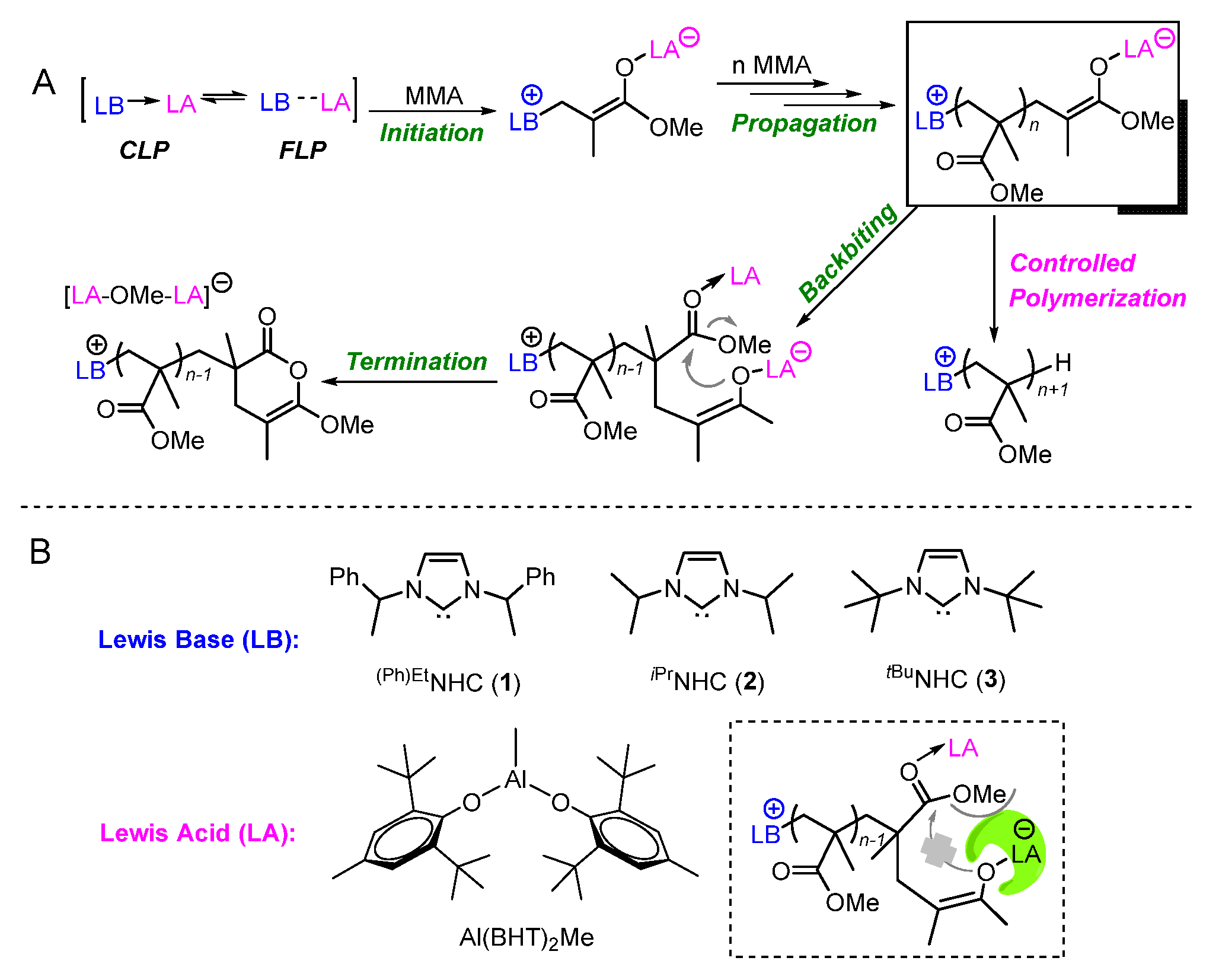
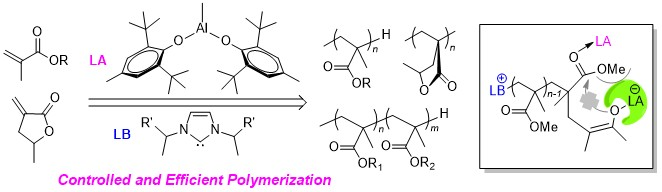
2. Results and Discussion
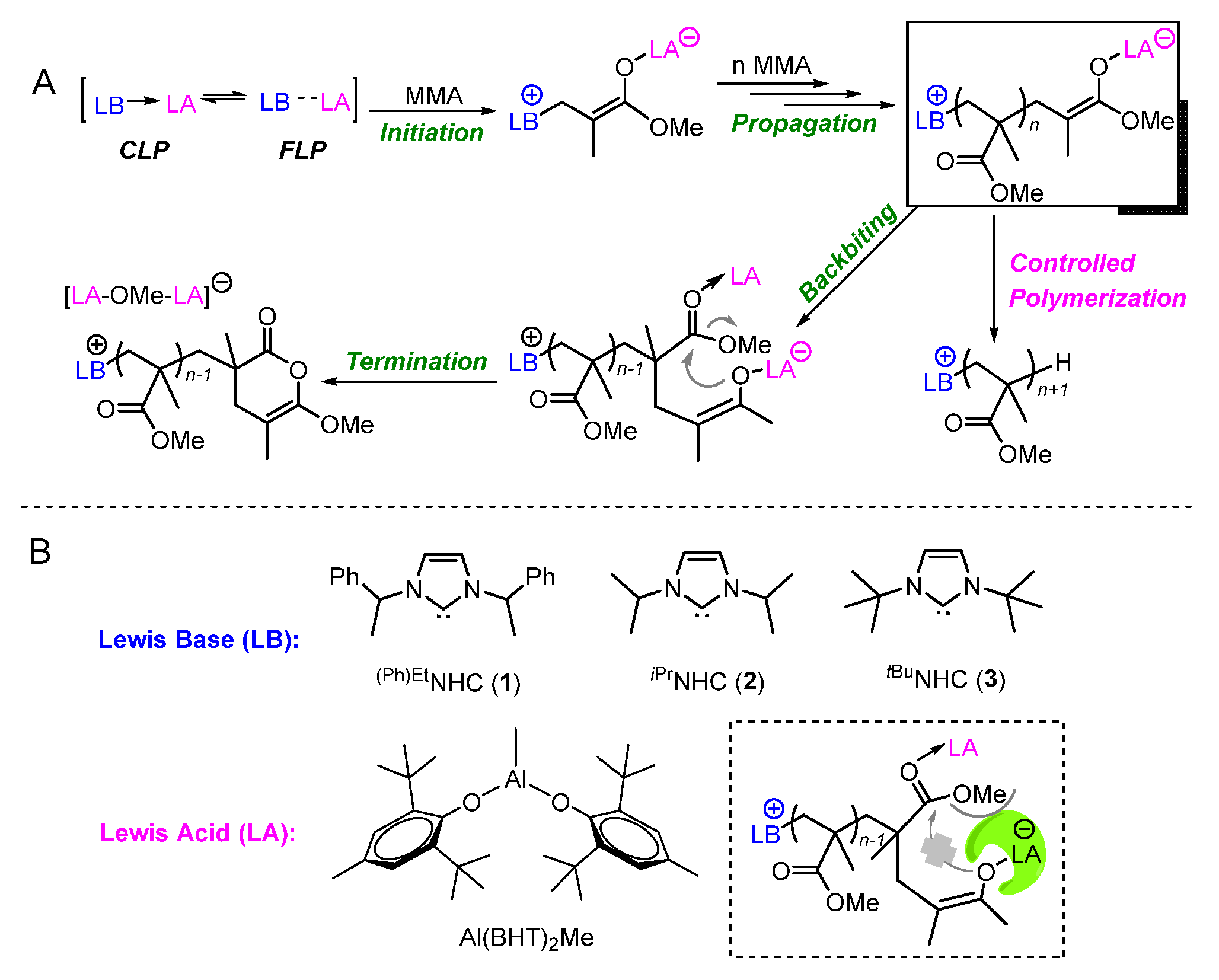
2.1. Interaction of Al(BHT)2Me with (Ph)EtNHC
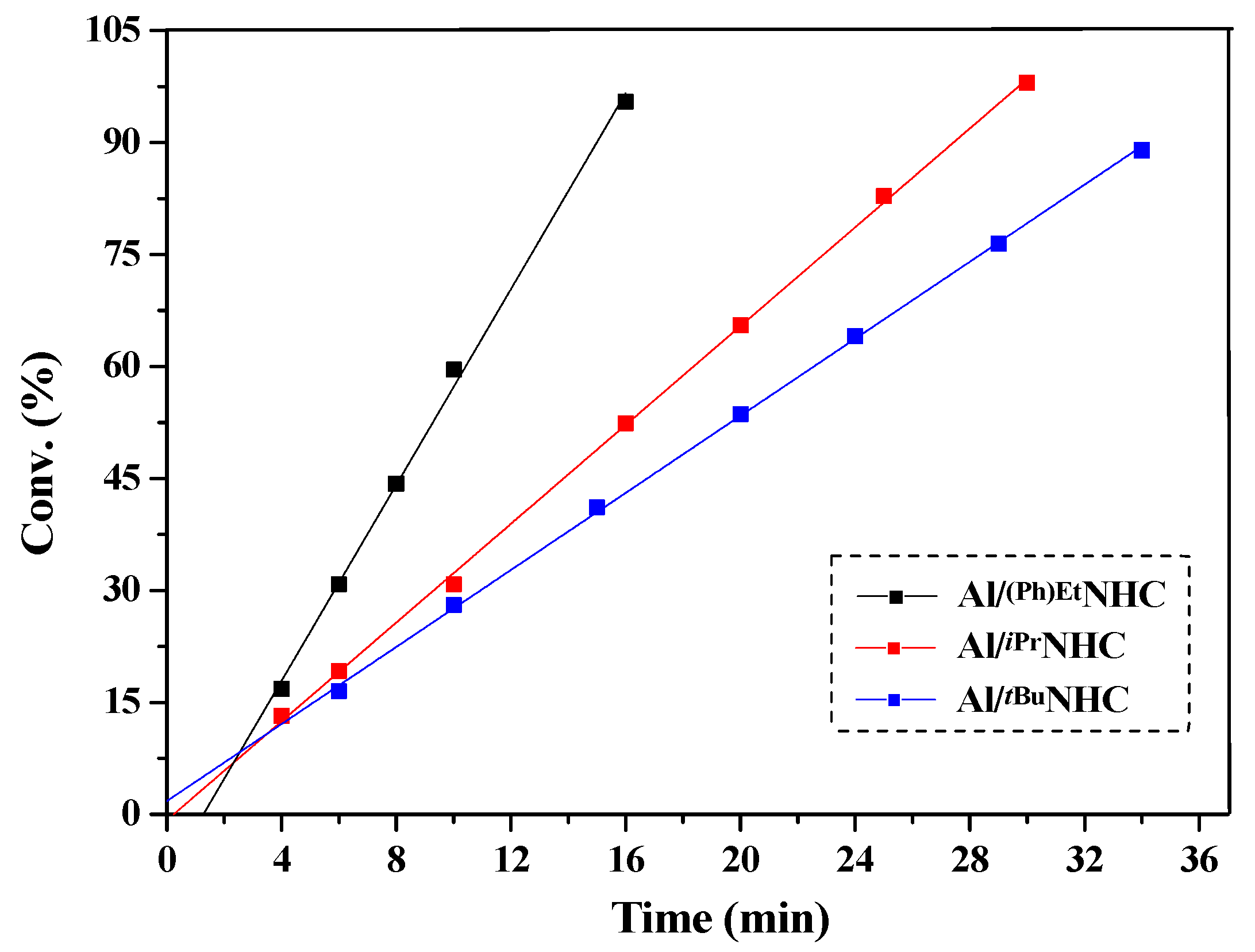
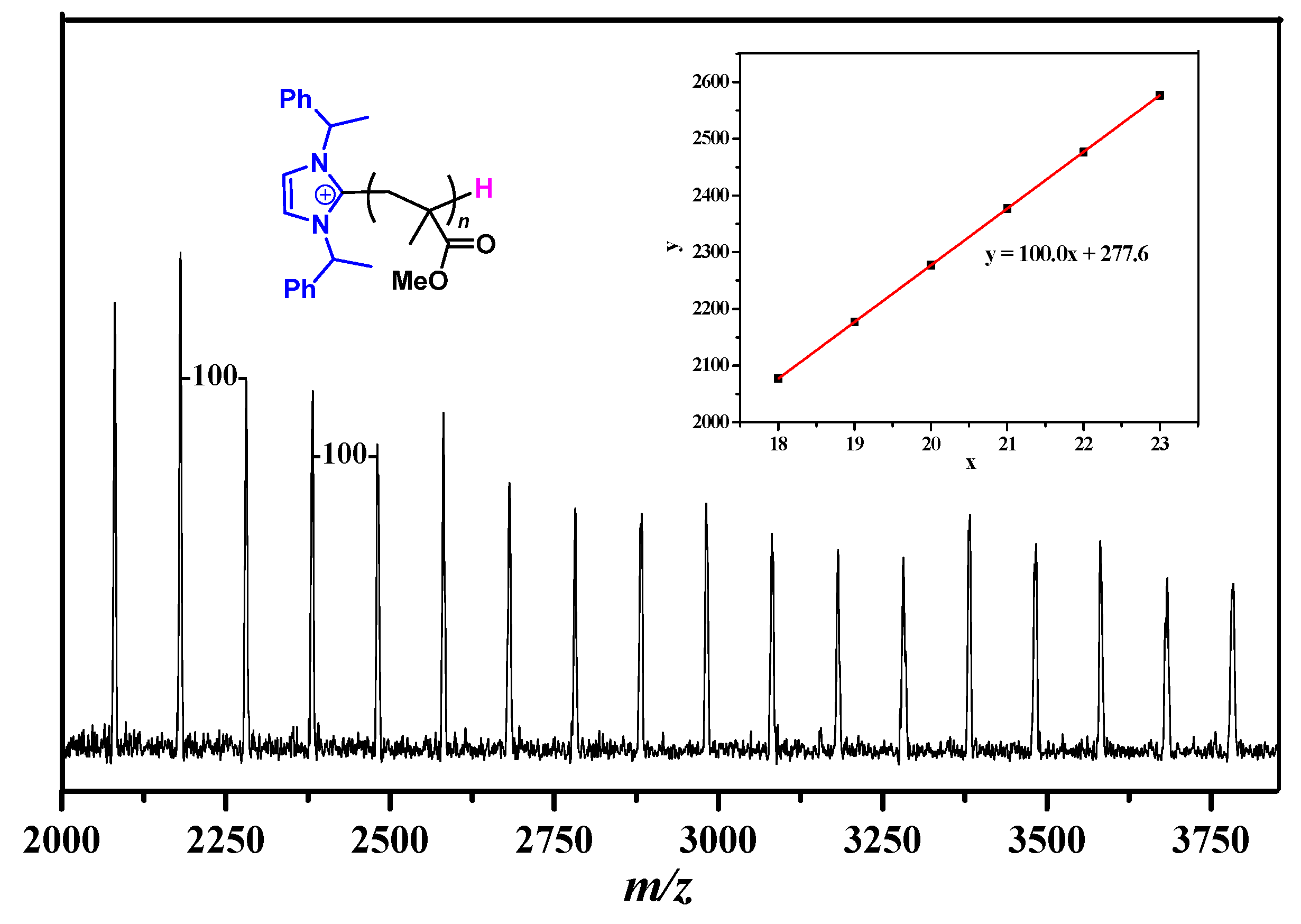
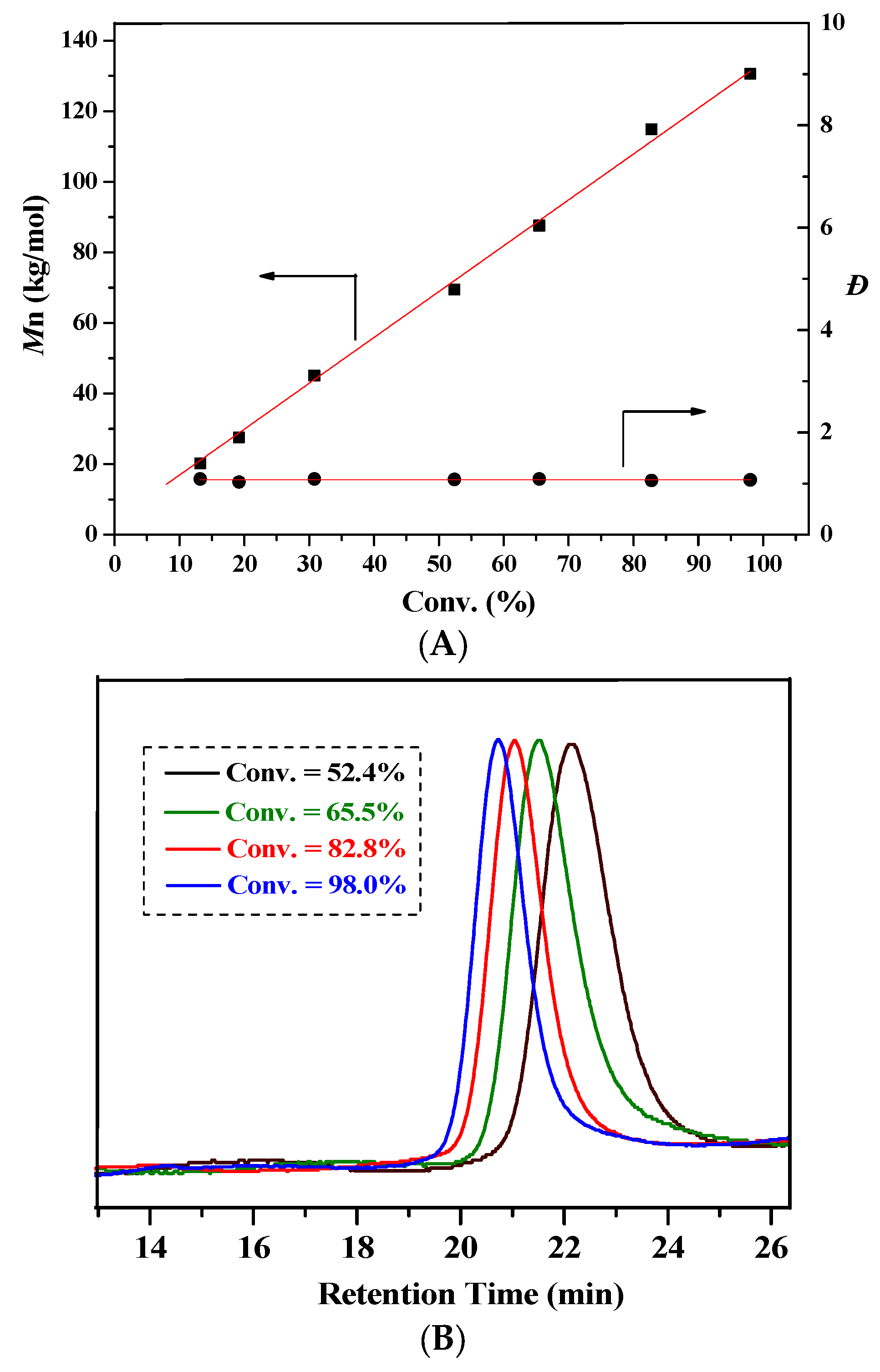
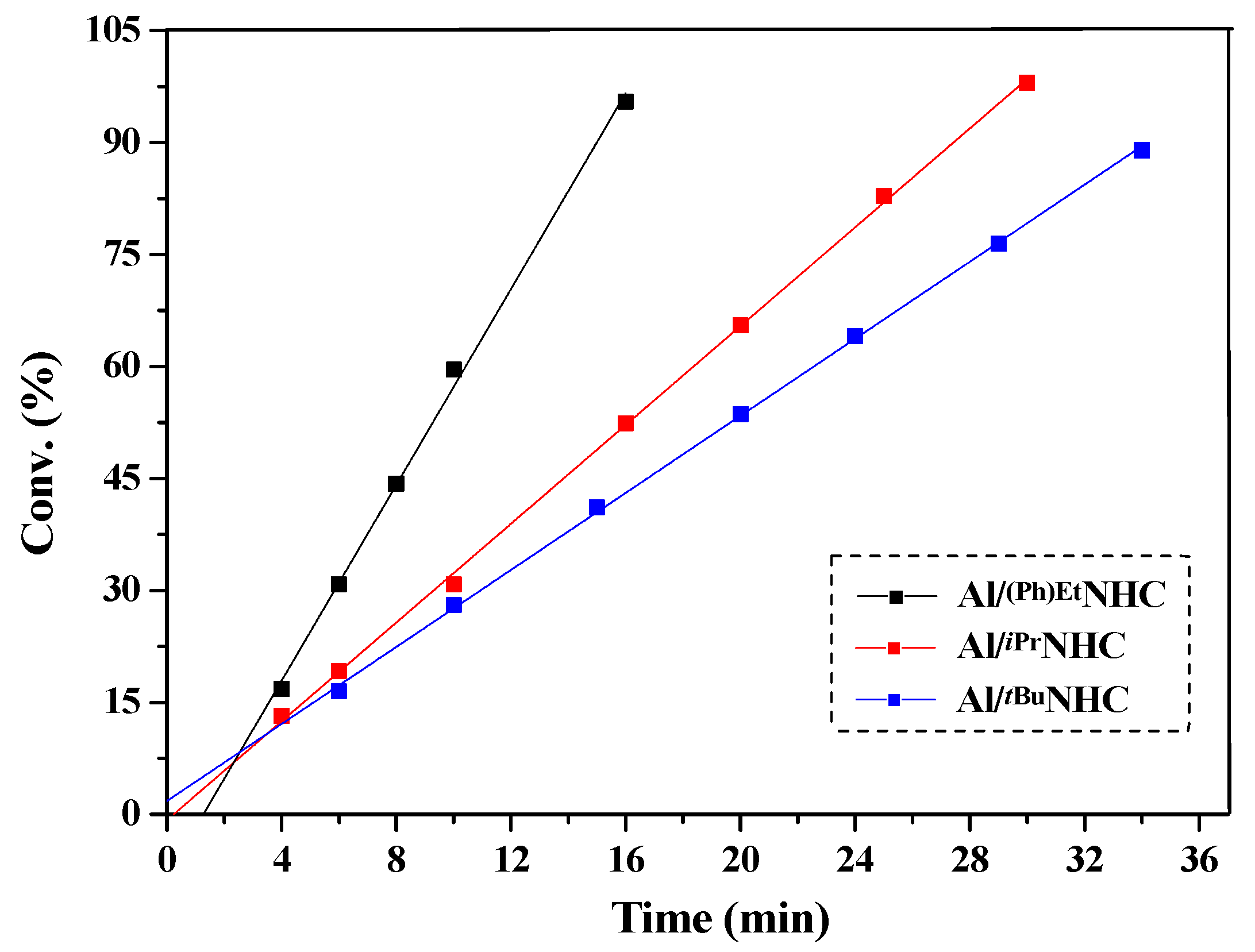
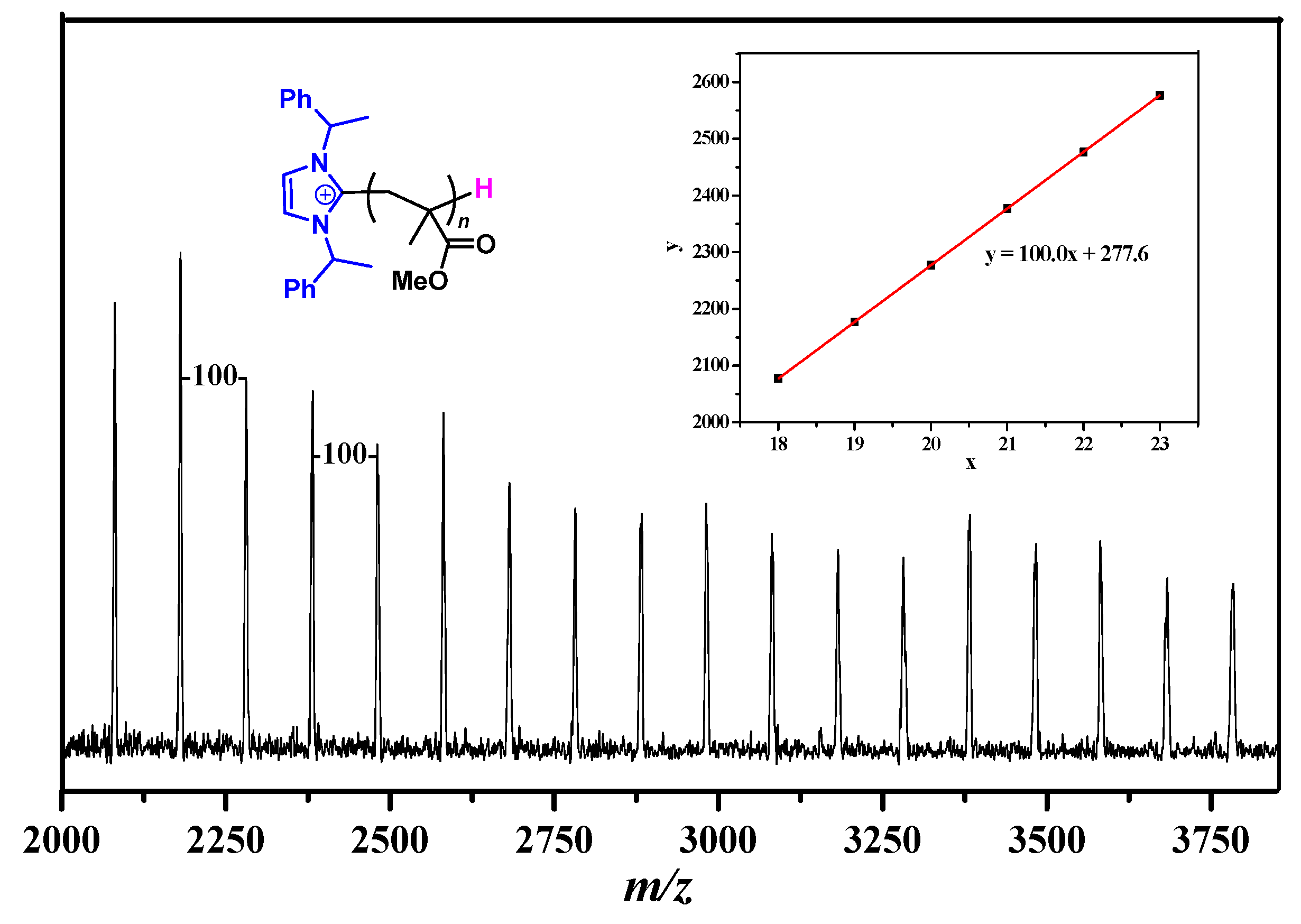
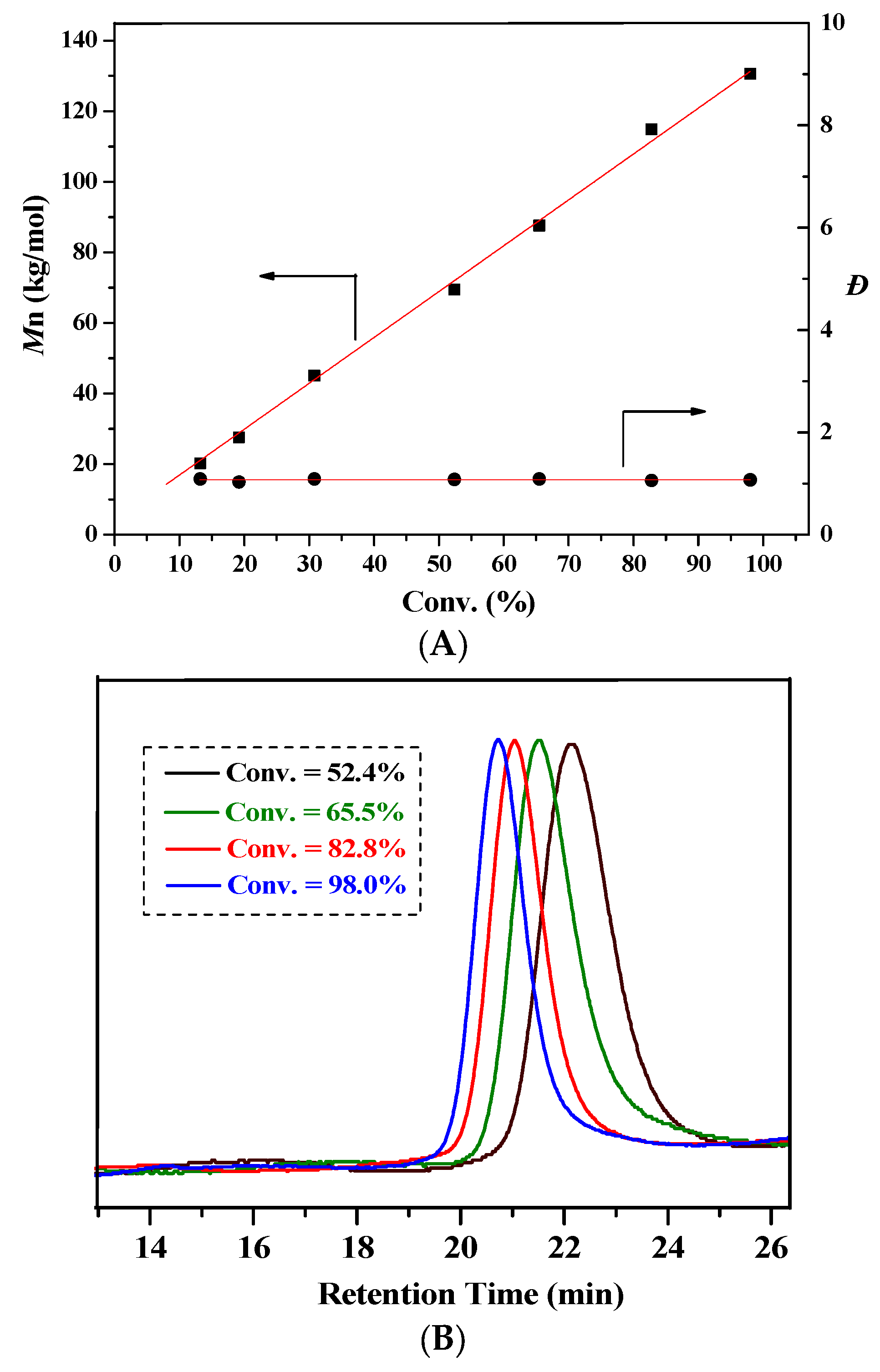
2.2. Characteristic of Polymerization by NHC/Al(BHT)2Me
3. Experimental Section
3.1. Materials, Reagents, and Methods
3.2. Stoichiometric Reaction of Al(BHT)2Me with MMA and In Situ Generation of MMA→Al(BHT)2Me Adduct
3.3. Stoichiometric Reaction of Al(BHT)2Me with (Ph)EtNHC and In Situ Generation of (Ph)EtNHC→Al(BHT)2Me Adduct
3.4. General Polymerization Procedures
3.5. Polymer Characterizations
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Note
- Welch, G.C.; Juan, R.R.S.; Masuda, J.D.; Stephan, D.W. Reversible, metal-free hydrogen activation. Science 2006, 314, 1124–1126. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W.; Erker, G. Frustrated Lewis pairs: Metal-free hydrogen activation and more. Angew. Chem. Int. Ed. 2010, 49, 46–76. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W.; Erker, G. Frustrated Lewis pair chemistry: Development and perspectives. Angew. Chem. Int. Ed. 2015, 54, 6400–6441. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W. Frustrated Lewis pairs: From concept to catalysis. Acc. Chem. Res. 2015, 48, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Feng, X.Q.; Du, H.F. Frustrated Lewis pairs catalyzed asymmetric metal-free hydrogenations and hydrosilylations. Acc. Chem. Res. 2018, 51, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.X. Polymerization by classical and frustrated Lewis pairs. Top. Curr. Chem. 2013, 334, 239–260. [Google Scholar] [PubMed]
- TOF value was calculated by the [monomer]/[catalyst] ratio divided by the reaction time when the polymerization reached typically quantitative conversion or the highest conversion reported.
- Zhang, Y.T.; Miyake, G.M.; Chen, E.Y.X. Alane-based classical and frustrated Lewis pairs in polymer synthesis: Rapid polymerization of MMA and naturally renewable methylene butyrolactones into high-molecular-weight polymers. Angew. Chem. Int. Ed. 2010, 49, 10158–10162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.T.; Miyake, G.M.; John, M.G.; Falivene, L.; Caporaso, L.; Cavallo, L.; Chen, E.Y.X. Lewis pair polymerization by classical and frustrated Lewis pairs: Acid, base and monomer scope and polymerization mechanism. Dalton Trans. 2012, 41, 9119–9134. [Google Scholar] [CrossRef] [PubMed]
- He, J.H.; Zhang, Y.T.; Chen, E.Y.X. Synthesis of pyridine- and 2-oxazoline-functionalized vinyl polymers by alane-based frustrated Lewis pairs. Synlett 2014, 25, 1534–1538. [Google Scholar]
- Chen, J.W.; Chen, E.Y.X. Lewis pair polymerization of acrylic monomers by N-heterocyclic carbenes and B(C6F5)3. Isr. J. Chem. 2015, 55, 216–225. [Google Scholar] [CrossRef]
- Chen, J.W.; Chen, E.Y.X. Reactivity of Amine/E(C6F5)3(E = B, Al) Lewis Pairs toward Linear and Cyclic Acrylic Monomers: Hydrogenation vs. Polymerization. Molecules 2015, 20, 9575–9590. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.Q.; Chen, E.Y.X. Probing site cooperativity of frustrated phosphine/borane Lewis pairs by a polymerization study. J. Am. Chem. Soc. 2014, 136, 1774–1777. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.B.; Ren, W.M.; Liu, S.J.; Xu, T.Q.; Wang, Y.B.; Lu, X.B. Controlled divinyl monomer polymerization mediated by Lewis pairs: A powerful synthetic strategy for functional polymers. ACS Macro Lett. 2014, 3, 896–899. [Google Scholar] [CrossRef]
- Xu, P.; Yao, Y.; Xu, X. Frustrated Lewis pair-like reactivity of rare-earth metal complexes: 1,4-addition reactions and polymerizations of conjugated polar alkenes. Chem. Eur. J. 2017, 23, 1263–1267. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.F.; Xu, X. Homoleptic rare-earth aryloxide based Lewis pairs for polymerization of conjugated polar alkenes. ACS Catal. 2018, 8, 198–202. [Google Scholar] [CrossRef]
- Piedra-Arroni, E.; Ladaviere, C.; Amgoune, A.; Bourissou, D. Ring-opening polymerization with Zn(C6F5)2-based Lewis pairs: Original and efficient approach to cyclic polyesters. J. Am. Chem. Soc. 2013, 135, 13306–13309. [Google Scholar] [CrossRef] [PubMed]
- Naumann, S.; Schmidt, F.G.; Frey, W.; Buchmeiser, M.R. Protected N-heterocyclic carbenes as latent pre-catalysts for the polymerization of ε-caprolactone. Polym. Chem. 2013, 4, 4172–4181. [Google Scholar] [CrossRef]
- Naumann, S.; Scholten, P.B.V.; Wilson, J.A.; Dove, A.P. Dual catalysis for selective ring-opening polymerization of lactones: Evolution toward simplicity. J. Am. Chem. Soc. 2015, 137, 14439–14445. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.B.; Chen, E.Y.X. From meso-lactide to isotactic polylactide: Epimerization by B/N Lewis pairs and kinetic resolution by organic catalysts. J. Am. Chem. Soc. 2015, 137, 12506–12509. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Wang, B.; Ji, H.Y.; Li, Y.S. Insights into the mechanism for ring-opening polymerization of lactide catalyzed by Zn(C6F5)2/organic superbase Lewis pairs. Catal. Sci. Technol. 2016, 6, 7763–7772. [Google Scholar] [CrossRef]
- Wang, Q.Y.; Zhao, W.C.; He, J.H.; Zhang, Y.T.; Chen, E.Y.X. Living ring-opening polymerization of lactones by N-heterocyclic olefin/Al(C6F5)3 Lewis pairs: Structures of intermediates, kinetics, and mechanism. Macromolecules 2016, 50, 123–136. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Nie, Y.Z.; Zhi, X.M.; Du, H.F.; Yang, J. Controlled ring-opening polymerization of α-amino acid N-carboxy-anhydride by frustrated amine/borane Lewis pairs. Chem. Commun. 2017, 53, 5155–5158. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.Y.; Wang, B.; Pan, L.; Li, Y.S. Lewis pairs for ring-opening alternating copolymerization of cyclic anhydrides and epoxides. Green Chem. 2018, 20, 641–648. [Google Scholar] [CrossRef]
- Yang, J.L.; Wu, H.L.; Li, Y.; Zhang, X.H.; Darensbourg, D.J. Perfectly alternating and regioselective copolymerization of carbonyl sulfide and epoxides by metal-free Lewis pairs. Angew. Chem. Int. Ed. 2017, 56, 5774–5779. [Google Scholar] [CrossRef] [PubMed]
- Grubbs, R.B.; Grubbs, R.H. 50th Anniversary perspective: Living polymerization—Emphasizing the molecule in macromolecules. Macromolecules 2017, 50, 6979–6997. [Google Scholar] [CrossRef]
- Aoshima, S.; Kanaoka, S. A renaissance in living cationic polymerization. Chem. Rev. 2009, 109, 5245–5287. [Google Scholar] [CrossRef] [PubMed]
- Braunecker, W.A.; Matyjaszewski, K. Controlled/living radical polymerization: Features, developments, and perspectives. Prog. Polym. Sci. 2007, 32, 93–146. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Xia, J. Atom transfer radical polymerization. Chem. Rev. 2001, 101, 2921–2990. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, M.; Terashima, T.; Sawamoto, M. Transition metal-catalyzed living radical polymerization: Toward perfection in catalysis and precision polymer synthesis. Chem. Rev. 2009, 109, 4963–5050. [Google Scholar] [CrossRef] [PubMed]
- Hadjichristidis, N.; Pitsikalis, M.; Pispas, S.; Iatrou, H. Polymers with complex architecture by living anionic polymerization. Chem. Rev. 2001, 101, 3747–3792. [Google Scholar] [CrossRef] [PubMed]
- Coates, G.W.; Hustad, P.D.; Reinartz, S. Catalysts for the living insertion polymerization of alkenes: Access to new polyolefin architectures using Ziegler–Natta chemistry. Angew. Chem. Int. Ed. 2002, 41, 2236–2257. [Google Scholar] [CrossRef]
- He, J.H.; Zhang, Y.T.; Falivene, L.; Caporaso, L.; Cavallo, L.; Chen, E.Y.X. Chain propagation and termination mechanisms for polymerization of conjugated polar alkenes by [Al]-Based frustrated Lewis pairs. Macromolecules 2014, 47, 7765–7774. [Google Scholar] [CrossRef]
- Jia, Y.B.; Wang, Y.B.; Ren, W.M.; Xu, T.Q.; Wang, J.; Lu, X.B. Mechanistic aspects of initiation and deactivation in N-heterocyclic olefin mediated polymerization of acrylates with alane as activator. Macromolecules 2014, 47, 1966–1972. [Google Scholar] [CrossRef]
- Ning, Y.; Chen, E.Y.X. Diastereospecific ion-pairing polymerization of functionalized alkenes by metallocene/Lewis acid hybrid catalysts. Macromolecules 2006, 39, 7204–7215. [Google Scholar] [CrossRef]
- Knaus, M.G.M.; Giuman, M.M.; Pöthig, A.; Rieger, B. End of frustration: Catalytic precision polymerization with highly interacting Lewis pairs. J. Am. Chem. Soc. 2016, 138, 7776–7781. [Google Scholar] [CrossRef] [PubMed]
- Ottou, W.N.; Conde-Mendizabal, E.; Pascual, A.; Wirotius, A.-L.; Bourichon, D.; Vignolle, J.; Robert, F.; Landais, Y.; Sotiropoulos, J.-M.; Miqueu, K.; et al. Organic Lewis pairs based on phosphine and electrophilic silane for the direct and controlled polymerization of methyl methacrylate: Experimental and theoretical investigations. Macromolecules 2017, 50, 762–774. [Google Scholar] [CrossRef]
- Allen, R.D.; Long, T.E.; Mcgrath, J.E. Preparation of high-purity, anionic-polymerization grade alkyl methacrylate monomers. Polym. Bull. 1986, 15, 127–134. [Google Scholar] [CrossRef]
- Shreve, A.P.; Mulhaupt, R.; Fultz, W.; Calabrese, J.; Robbins, W.; Ittel, S. Sterically hindered aryloxide-substituted alkylaluminum compounds. Organometallics 1988, 7, 409–416. [Google Scholar] [CrossRef]
- Feng, S.; Roof, G.R.; Chen, E.Y.X. Tantalum(V)-based metallocene, half-metallocene, and non-metallocene complexes as ethylene-1-octene copolymerization and methyl methacrylate polymerization catalysts. Organometallics 2002, 21, 832–839. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Goossen, L.J.; Artus, G.R.J.; Köcher, C. Complexes of chiral imidazolin-2-ylidene ligands. Organometallics 1997, 16, 2472–2477. [Google Scholar] [CrossRef]
- Kuhn, N.; Kratz, T. Synthesis of imidazol-2-ylidenes by reduction of imidazole-2(3H)-thiones. Synthesis 1993, 6, 561–562. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run | LB | LA | Monomer (M) | M/LA/LB | Solvent | Time (min) | Conv. (%) 2 | Mn 3 (kg/mol) | Đ 3 | I* 4 (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | - | MMA | 100/-/1 | TOL | 1440 | 0 | - | - | - |
| 2 | - | Al(BHT)2Me | MMA | 100/1/- | TOL | 1440 | 0 | - | - | - |
| 3 | 1 | Al(BHT)2Me | MMA | 100/2/1 | TOL | 3 | 100 | 54.1 | 1.11 | 19.0 |
| 4 5 | 1 | Al(BHT)2Me | MMA | 100/2/1 | TOL | 30 | 100 | 343.2 | 1.14 | 3.00 |
| 5 | 1 | Al(BHT)2Me | MMA | 500/2/1 | TOL | 16 | 100 | 102.7 | 1.06 | 50.0 |
| 6 | 1 | Al(BHT)2Me | MMA | 500/3/1 | TOL | 10 | 100 | 82.5 | 1.13 | 60.6 |
| 7 | 1 | Al(BHT)2Me | MMA | 500/2/1 | DCM | 25 | 100 | 127.9 | 1.07 | 39.1 |
| 8 | 1 | B(C6F5)3 | MMA | 100/2/1 | TOL | 75 | 99 | 15.8 | 1.48 | 65.2 |
| 9 | 1 | Al(C6F5)3 | MMA | 100/2/1 | TOL | 1440 | 0 | - | - | - |
| 10 | 1 | AlMe3 | MMA | 100/2/1 | TOL | 1440 | 0 | - | - | - |
| 11 | 1 | AlEt3 | MMA | 100/2/1 | TOL | 1440 | 19.6 | - | - | - |
| 12 | 1 | AliBu3 | MMA | 100/2/1 | TOL | 1440 | 99.8 | 106 | 1.47 | 9.70 |
| 13 | 2 | Al(BHT)2Me | MMA | 500/2/1 | TOL | 30 | 98 | 130.6 | 1.07 | 38.0 |
| 14 | 3 | Al(BHT)2Me | MMA | 250/2/1 | TOL | 34 | 93.2 | 258.0 6 | 1.08 6 | 9.03 |
| 15 | 1 | Al(BHT)2Me | nBuMA | 500/2/1 | TOL | 20 | 100 | 128.9 | 1.18 | 55.0 |
| 16 | 1 | Al(BHT)2Me | MMBL | 500/2/1 | DCM+ TOL 7 | 40 | 98.5 | 133.0 | 1.04 | 41.5 |
| 17 8 | 1 | Al(BHT)2Me | MMA/nBuMA | (400 + 250)/2/1 | TOL | 16 10 | 100 | 87.4 105.5 | 1.13 1.09 | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Zhang, Y.; Hong, M. Controlled and Efficient Polymerization of Conjugated Polar Alkenes by Lewis Pairs Based on Sterically Hindered Aryloxide-Substituted Alkylaluminum. Molecules 2018, 23, 442. https://doi.org/10.3390/molecules23020442
Wang X, Zhang Y, Hong M. Controlled and Efficient Polymerization of Conjugated Polar Alkenes by Lewis Pairs Based on Sterically Hindered Aryloxide-Substituted Alkylaluminum. Molecules. 2018; 23(2):442. https://doi.org/10.3390/molecules23020442
Chicago/Turabian StyleWang, Xiaojun, Yixin Zhang, and Miao Hong. 2018. "Controlled and Efficient Polymerization of Conjugated Polar Alkenes by Lewis Pairs Based on Sterically Hindered Aryloxide-Substituted Alkylaluminum" Molecules 23, no. 2: 442. https://doi.org/10.3390/molecules23020442
APA StyleWang, X., Zhang, Y., & Hong, M. (2018). Controlled and Efficient Polymerization of Conjugated Polar Alkenes by Lewis Pairs Based on Sterically Hindered Aryloxide-Substituted Alkylaluminum. Molecules, 23(2), 442. https://doi.org/10.3390/molecules23020442