Chemoselective Polymerization of Polar Divinyl Monomers with Rare-Earth/Phosphine Lewis Pairs


Abstract
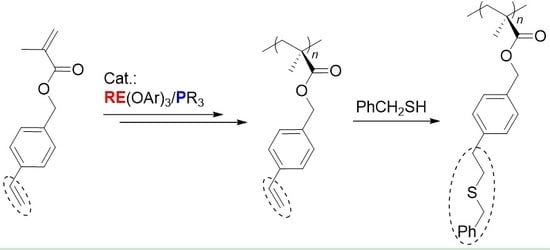
:
1. Introduction
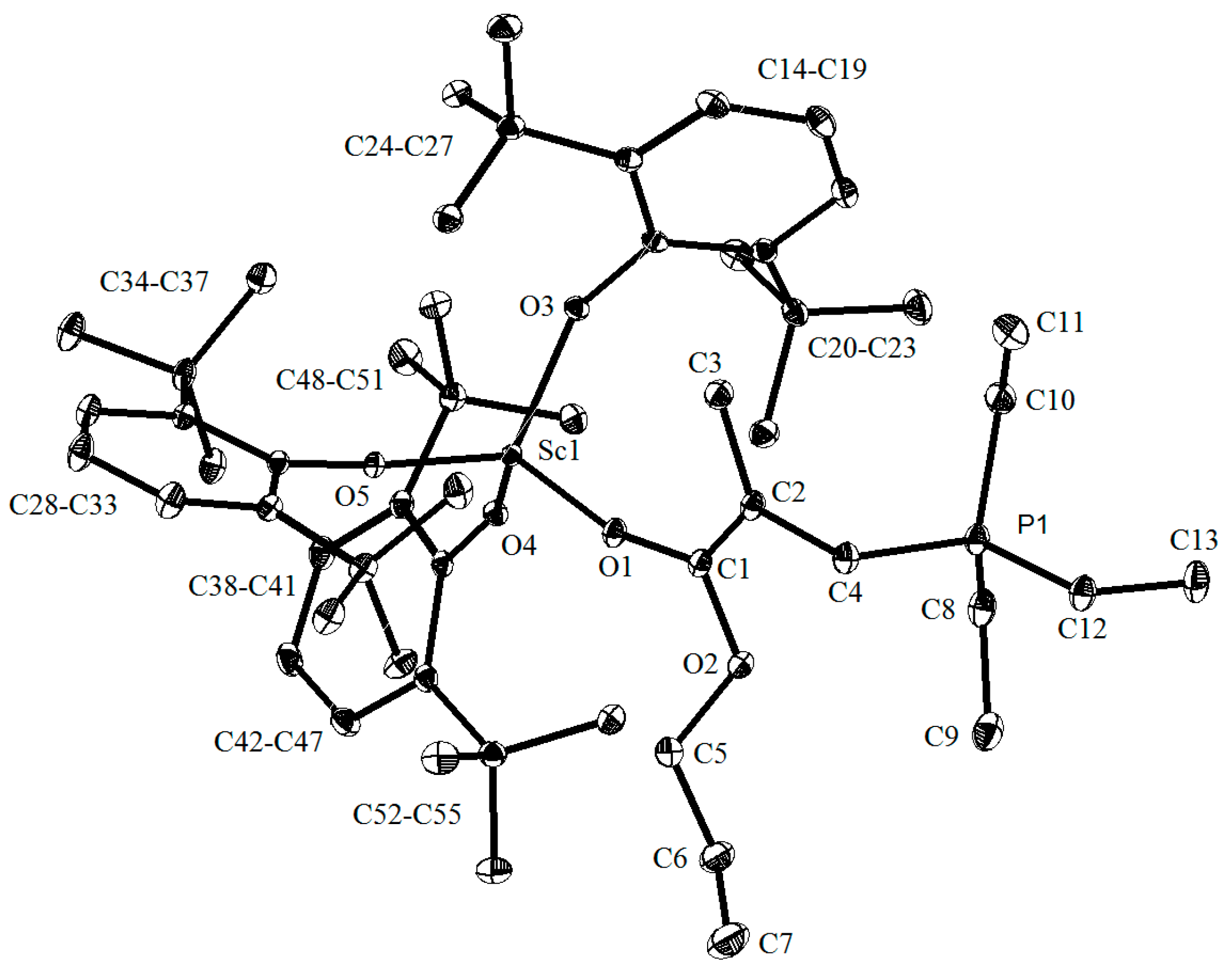
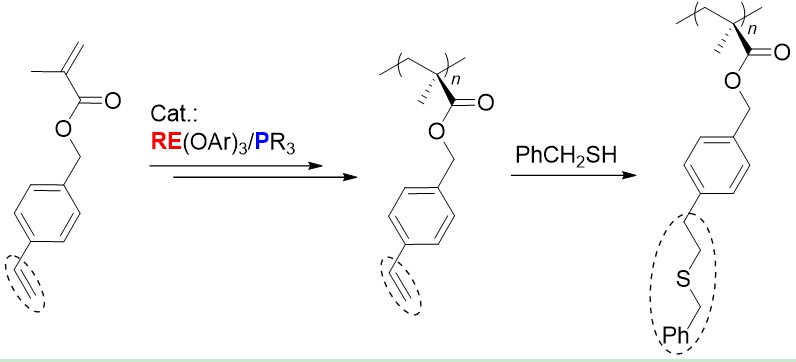
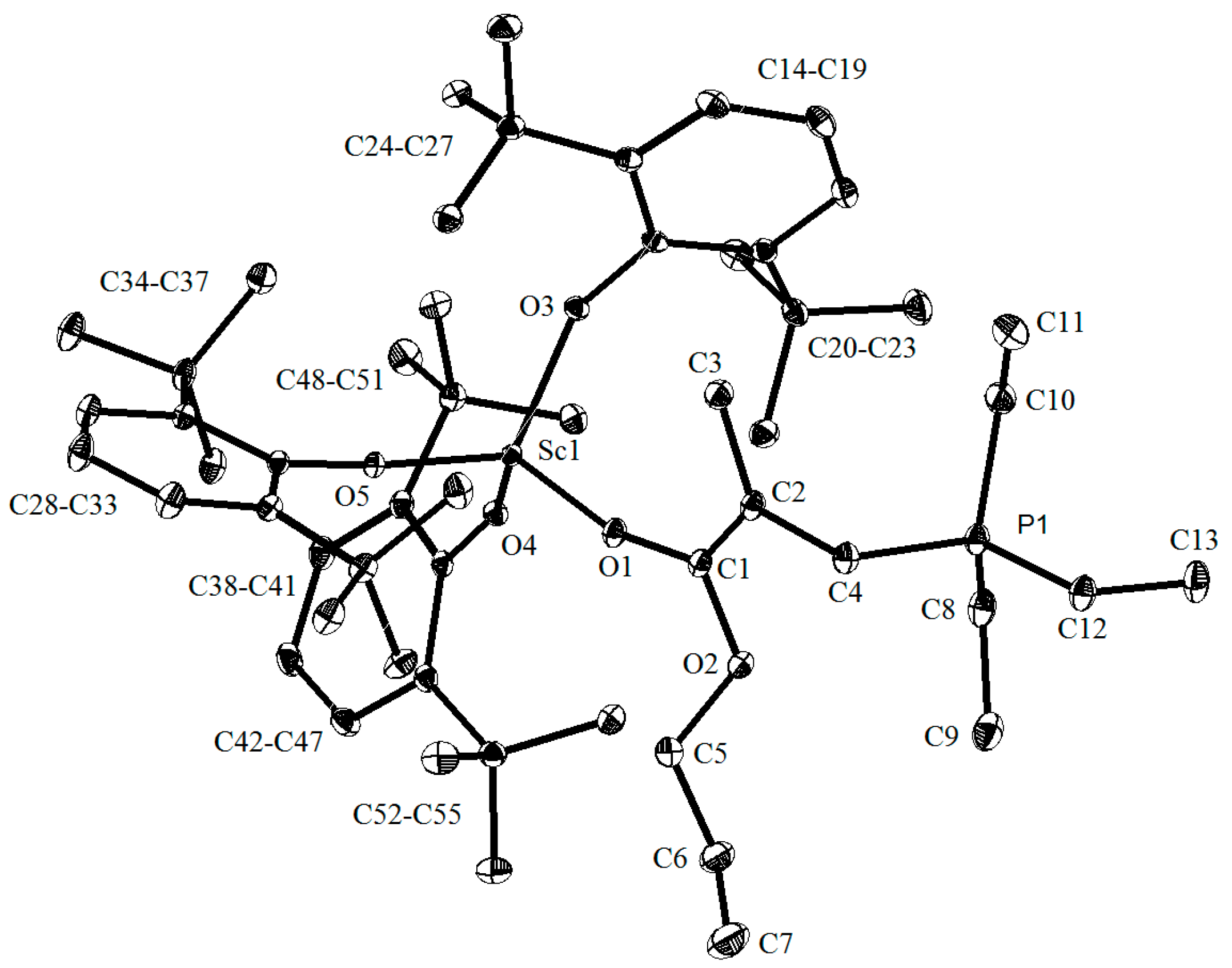
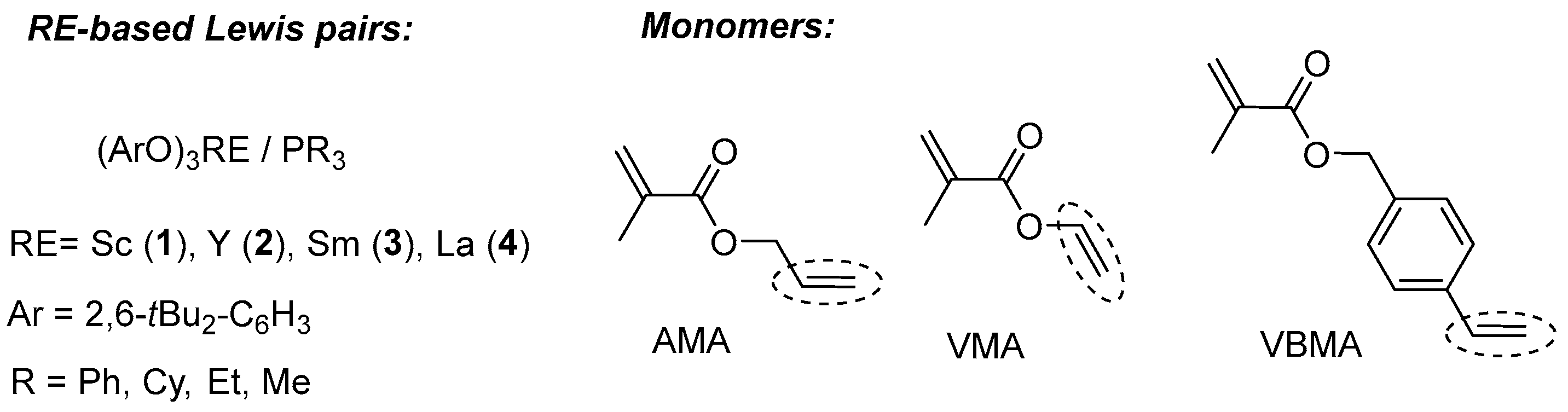
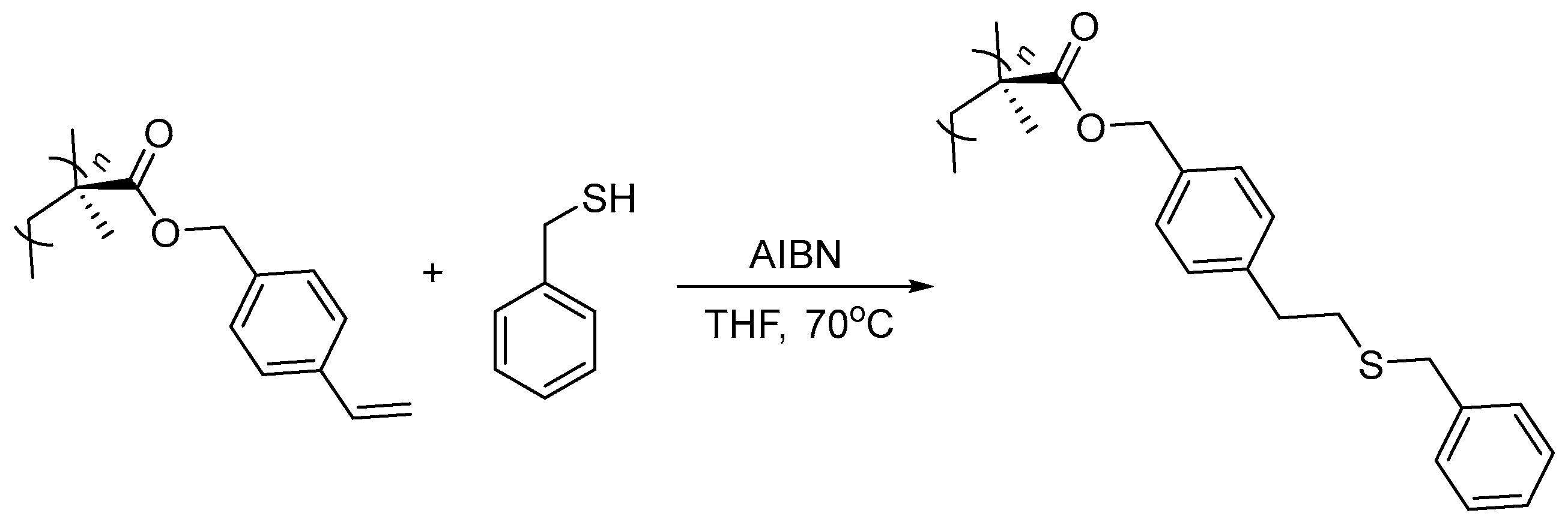
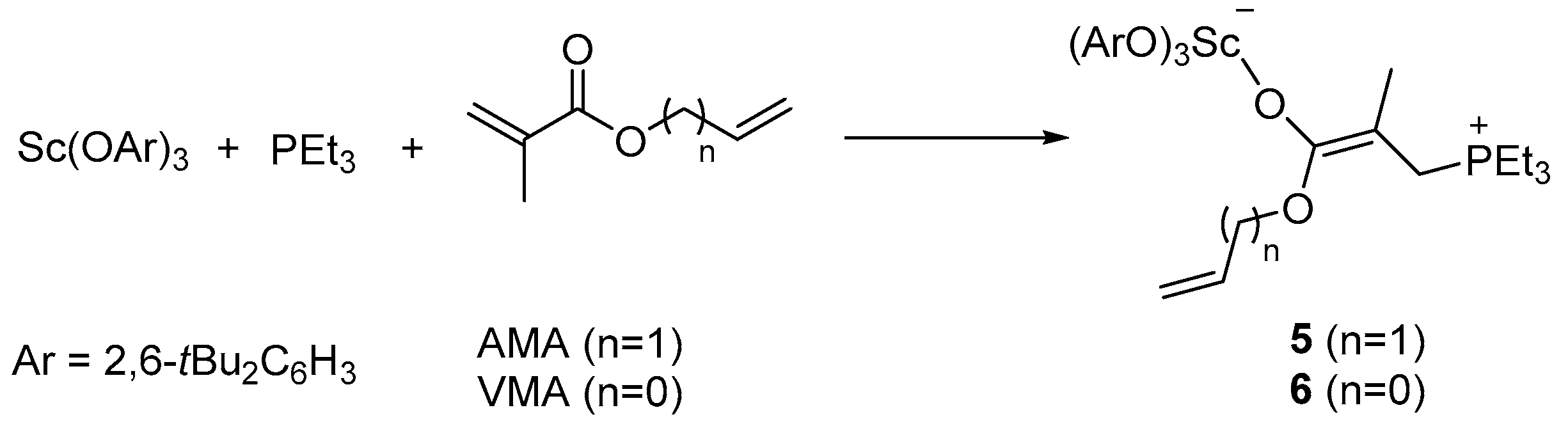
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Procedures and Compound Characterization
3.2.1. Preparation and Characterization of Complex 5
3.2.2. Preparation and Characterization of Complex 6
3.2.3. General Polymerization Procedures
3.2.4. Polymer Characterizations
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Note
- Cherian, A.E.; Sun, F.C.; Sheiko, S.S.; Coates, G.W. Formation of nanoparticles by intramolecular cross-linking: Following the reaction progress of single polymer chains by atomic force microscopy. J. Am. Chem. Soc. 2007, 129, 11350–11351. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Hempenius, M.A.; Vancso, G.J. Redox-active cross-linkable poly(ionic liquid)s. J. Am. Chem. Soc. 2012, 134, 4023–4025. [Google Scholar] [CrossRef] [PubMed]
- Wendland, M.S.; Zimmerman, S.C. Synthesis of cored dendrimers. J. Am. Chem. Soc. 1999, 121, 1389–1390. [Google Scholar] [CrossRef]
- Powell, K.T.; Cheng, C.; Wooley, K.L. Complex amphiphilic hyperbranched fluoropolymers by atom transfer radical self-condensing vinyl (co)polymerization. Macromolecules 2007, 40, 4509–4515. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chen, P.; Deng, C.; Meng, F.; Cheng, R.; Zhong, Z. A simple and versatile synthetic strategy to functional polypeptides via vinyl sulfone-substituted L-cysteine N-carboxyanhydride. Macromolecules 2013, 46, 6723–6730. [Google Scholar] [CrossRef]
- Stevens, D.M.; Tempelaar, S.; Dove, A.P.; Harth, E. Nanosponge formation from organocatalytically synthesized poly(carbonate) copolymers. ACS Macro Lett. 2012, 1, 915–918. [Google Scholar] [CrossRef] [PubMed]
- Coates, G.W.; Grubbs, R.H. Quantitative ring-closing metathesis of polyolefins. J. Am. Chem. Soc. 1996, 118, 229–230. [Google Scholar] [CrossRef]
- Ishizone, T.; Uehara, G.; Hirao, A.; Nakahama, S. Anionic polymerization of monomers containing functional groups. 13. Anionic polymerizations of 2-, 3- and 4-(3,3-dimethyl-1-butynyl)styrenes, 2-, 3-, and 4-(1-hexynyl)styrenes, and 4-(phenylethynyl)styrene. Macromolecules 1998, 31, 3764–3774. [Google Scholar] [CrossRef]
- Tanaka, S.; Goseki, R.; Ishizone, T.; Hirao, A. Synthesis of well-defined novel reactive block polymers containing a poly(1,4-divinylbenzene) segment by living anionic polymerization. Macromolecules 2014, 47, 2333–2339. [Google Scholar] [CrossRef]
- Pariś, R.; de la Fuente, J.L. Bulk atom transfer radical polymerization of allyl methacrylate. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 2395–2406. [Google Scholar] [CrossRef]
- Pariś, R.; de la Fuente, J.L. Solvent effect on the atom transfer radical polymerization of allyl methacrylate. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 6247–6261. [Google Scholar] [CrossRef]
- Nagelsdiek, R.; Mennicken, M.; Maier, B.; Keul, H.; Hӧcker, H. Synthesis of polymers containing cross-linkable groups by atom transfer radical polymerization: Poly(allyl methacrylate) and copolymers of allyl methacrylate and styrene. Macromolecules 2004, 37, 8923–8932. [Google Scholar] [CrossRef]
- Sugiyama, F.; Satoh, K.; Kamigaito, M. Regiospecific radical polymerization of vinyl methacrylate in the presence of Lewis acids into soluble polymers with pendent vinyl ester substituents. Macromolecules 2008, 41, 3042–3048. [Google Scholar] [CrossRef]
- Vardareli, T.K.; Keskin, S.; Usanmaz, A. Synthesis and characterization of poly(allyl methacrylate) obtained by free radical initiator. J. Macromol. Sci. Part A Pure Appl. Chem. 2008, 45, 302–311. [Google Scholar] [CrossRef]
- Vidal, F.; Chen, E.Y.-X. Precision polymer synthesis via chemoselective, stereoselective, and living/controlled polymerization of polar divinyl monomers. Synlett 2017, 28, 1028–1039. [Google Scholar]
- Murali Mohan, Y.; Raghunadh, V.; Sivaram, S.; Baskaran, D. Reactive polymers bearing styrene pendants through selective anionic polymerization of 4-vinylbenzyl methacrylate. Macromolecules 2012, 45, 3387–3393. [Google Scholar] [CrossRef]
- Vidal, F.; Gowda, R.R.; Chen, E.Y.-X. Chemoselective, stereospecific, and living polymerization of polar divinyl monomers by chiral zirconocenium catalysts. J. Am. Chem. Soc. 2015, 137, 9469–9480. [Google Scholar] [CrossRef] [PubMed]
- Vidal, F.; Falivene, L.; Caporaso, L.; Cavallo, L.; Chen, E.Y.-X. Robust cross-linked stereocomplexes and C60 inclusion complexes of vinyl-functionalized stereoregular polymers derived from chemo/stereoselective coordination polymerization. J. Am. Chem. Soc. 2016, 138, 9533–9547. [Google Scholar] [CrossRef] [PubMed]
- Gowda, R.R.; Chen, E.Y.-X. Organocatalytic and chemoselective polymerization of multivinyl-functionalized γ-butyrolactones. ACS Macro Lett. 2016, 5, 772–776. [Google Scholar] [CrossRef]
- Xu, T.; Liu, J.; Lu, X.B. Highly active half-metallocene yttrium catalysts for living and chemoselective polymerization of allyl methacrylate. Macromolecules 2015, 48, 7428–7434. [Google Scholar] [CrossRef]
- Welch, G.C.; Juan, R.R.S.; Masuda, J.D.; Stephan, D.W. Reversible metal-free hydrogen activiation. Science 2006, 314, 1124–1126. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W.; Erker, G. Frustrated Lewis pair chemistry: Development and perspectives. Angew. Chem. Int. Ed. 2015, 54, 6400–6441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Miyake, G.M.; Chen, E.Y.-X. Alane-based classical and frustrated Lewis pairs in polymer synthesis: Rapid polymerization of MMA and naturally renewable methylene butyrolactones into high-molecular-weight polymers. Angew. Chem. Int. Ed. 2010, 49, 10158–10162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Miyake, G.M.; John, M.G.; Falivene, L.; Caporaso, L.; Cavallo, L.; Chen, E.Y.-X. Lewis pair polymerization by classical and frustrated Lewis pairs: Acid, base and monomer scope and polymerization mechanism. Dalton Trans. 2012, 41, 9119–9134. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Chen, E.Y.-X. Probing site cooperativity of frustrated phosphine/borane Lewis pairs by a polymerization study. J. Am. Chem. Soc. 2014, 136, 1774–1777. [Google Scholar] [CrossRef] [PubMed]
- Knaus, M.G.M.; Giuman, M.M.; Pӧthig, A.; Rieger, B. End of frustration: Catalytic precision polymerization with highly interacting Lewis pairs. J. Am. Chem. Soc. 2016, 138, 7776–7781. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.B.; Ren, W.M.; Liu, S.J.; Xu, T.; Wang, Y.B.; Lu, X.B. Controlled divinyl monomer polymerization mediated by Lewis pairs: A powerful synthetic strategy for functional polymers. ACS Macro Lett. 2014, 3, 896–899. [Google Scholar] [CrossRef]
- Chen, J.; Chen, E.Y.-X. Lewis pair polymerization of acrylic monomers by N-heterocyclic carbenes and B(C6F5)3. Isr. J. Chem. 2015, 55, 216–226. [Google Scholar] [CrossRef]
- Xu, P.; Yao, Y.; Xu, X. Frustrated Lewis pair-like reactivity of rare-earth metal complexes: 1,4-addition reactions and polymerizations of conjugated polar alkenes. Chem. A Eur. J. 2017, 23, 1263–1267. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Xu, X. Homoleptic rare-earth aryloxide based Lewis pairs for polymerization of conjugated polar alkenes. ACS Catal. 2018, 8, 198–202. [Google Scholar] [CrossRef]
- Arndt, S.; Spaniol, T.P.; Okuda, J. Homogeneous ethylene-polymerization catalysts based on alkyl cations of the rare-earth metals: Are dicationic mono(alkyl) complexes the active species? Angew. Chem. Int. Ed. 2003, 42, 5075–5079. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Marks, T.J. Organolanthanide-catalyzed hydroamination. Acc. Chem. Res. 2004, 37, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Greenley, R.Z. Polymer Handbook, 3rd ed.; Immergut, E.H., Brandup, J., Eds.; Wiley: New York, NY, USA, 1989; pp. 267–274. [Google Scholar]
- The divinyl monomers was regarded as a copolymerization system of two independent and competing vinyl monomers.
- Pugh, C.; Percec, V. Synthesis and group transfer polymerization and copolymerization of p-vinylbenzyl methacrylate. Polym. Bull. 1985, 14, 109–116. [Google Scholar] [CrossRef]
- Lappert, M.F.; Singh, A.; Smith, R.G. Hydrocarbon-soluble homoleptic bulky aryl oxides of the lanthanide metals [Ln(OArR)3]. Inorg. Synth. 1990, 27, 164–168. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |
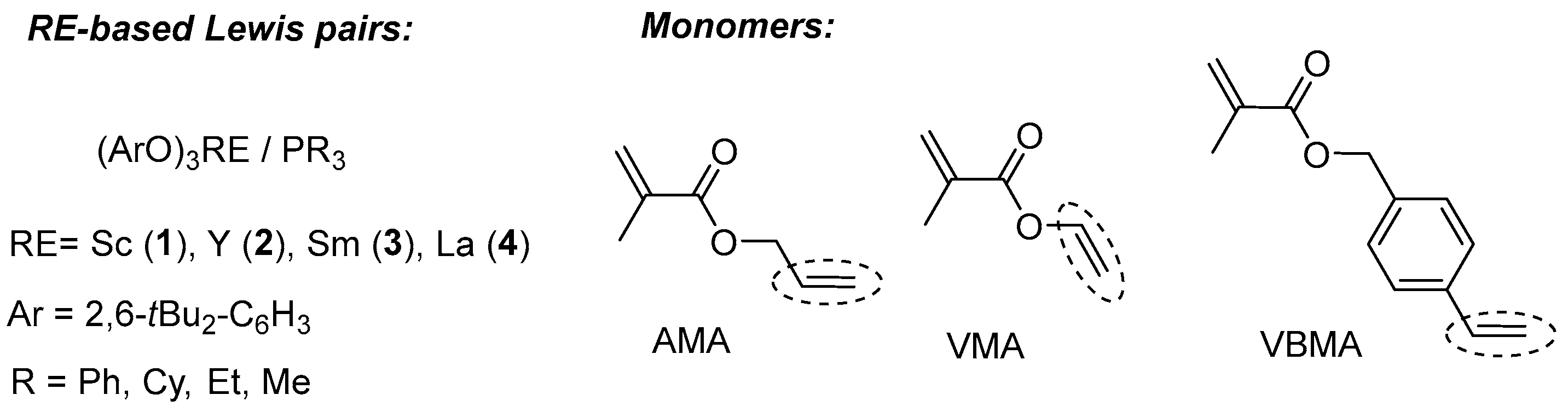
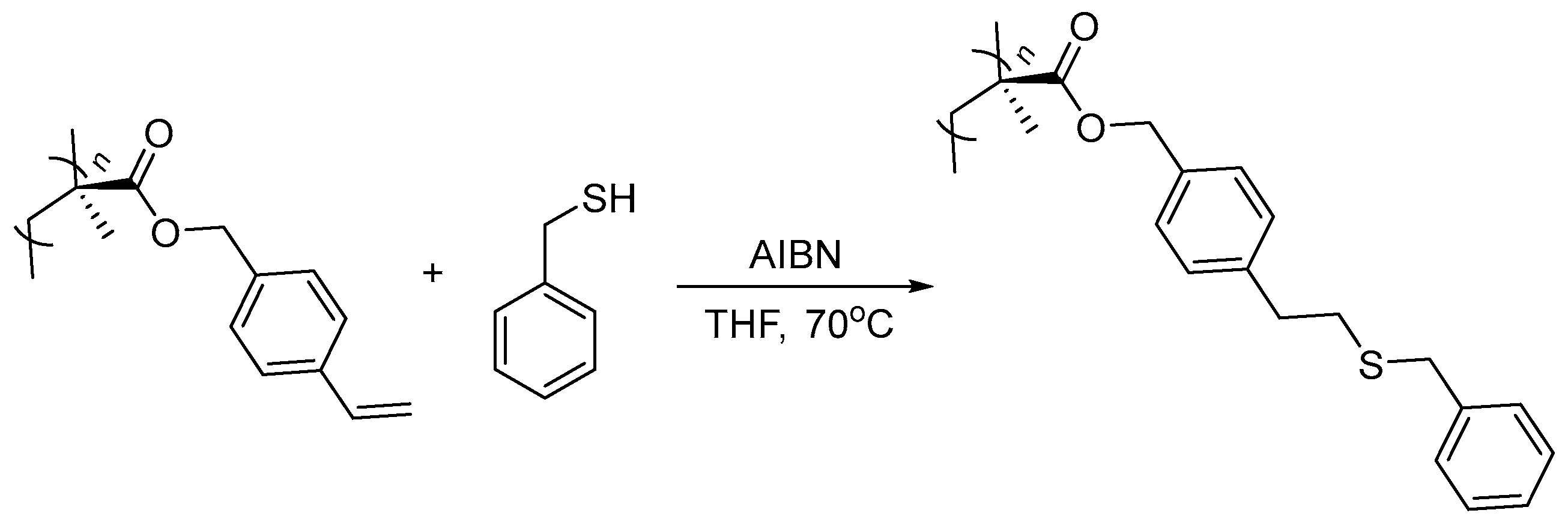
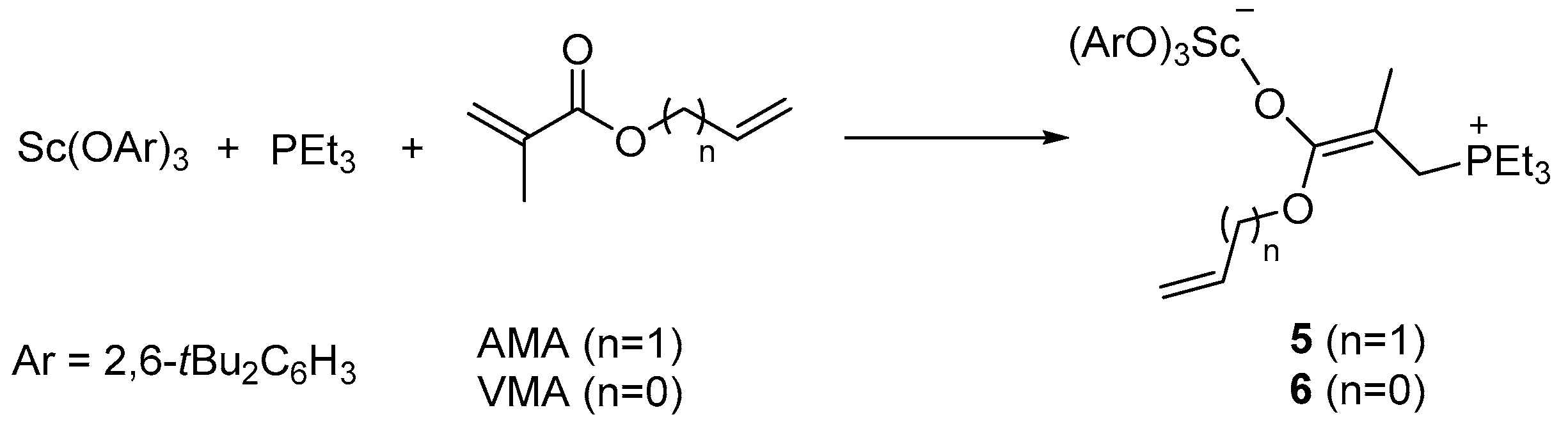

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Monomer | LA | LB | [M]/[LB] | T (min) | Conv. (%) | Mn (104 g/mol) | PDI (Mw/Mn) | Mn(theo) b (104 g/mol) | rr (%) | mr (%) | mm (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | AMA | 1 | PPh3 | 200 | 1440 | 0 | - | - | - | - | - | - |
| 2 | AMA | 1 | PCy3 | 200 | 25 | 100 | 12.3 | 1.33 | 2.55 | 79.8 | 19.6 | 0.6 |
| 3 | AMA | 1 | PEt3 | 200 | 15 | 100 | 5.03 | 1.23 | 2.54 | 79.9 | 19.5 | 0.6 |
| 4 c | AMA | 1 | PEt3 | 200 | 1440 | 75 | 10.8 | 1.38 | 2.54 | 87.8 | 11.4 | 0.8 |
| 5 | AMA | 1 | PMe3 | 200 | 30 | 90 | 5.09 | 1.23 | 2.53 | 79.2 | 20.0 | 0.8 |
| 6 | AMA | 2 | PEt3 | 200 | 3 | 100 | 8.57 | 1.41 | 2.54 | 75.3 | 23.5 | 1.2 |
| 7 | AMA | 3 | PEt3 | 200 | 1 | 100 | 9.16 | 1.34 | 2.54 | 75.0 | 23.7 | 1.7 |
| 8 | AMA | 4 | PEt3 | 200 | <1 | 100 | 10.0 | 1.42 | 2.54 | 72.5 | 26.0 | 1.5 |
| 9 | AMA | 4 | PEt3 | 400 | 5 | 100 | 15.3 | 1.76 | 5.06 | 72.8 | 25.4 | 1.8 |
| 10 | VMA | 1 | PEt3 | 100 | 10 | 100 | 5.62 | 1.36 | 1.13 | 75.6 | 22.6 | 1.8 |
| 11 | VMA | 1 | PMe3 | 100 | 10 | 100 | 7.59 | 1.31 | 1.13 | 76.8 | 21.6 | 1.6 |
| 12 | VMA | 4 | PEt3 | 100 | 1 | 100 | 4.74 | 1.76 | 1.13 | 69.3 | 29.1 | 1.6 |
| 13 | VBMA | 1 | PEt3 | 100 | 45 | 100 | 2.55 | 1.87 | 2.03 | 75.0 | 22.5 | 2.5 |
| 14 | VBMA | 1 | PMe3 | 100 | 45 | 100 | 2.42 | 1.99 | 2.03 | 76.2 | 22.8 | 1.0 |
| 15 | VBMA | 4 | PEt3 | 100 | 5 | 100 | 2.92 | 2.00 | 2.03 | 72.1 | 26.3 | 1.6 |
| Entry | Monomer | Cat. | LA | [M]/[Cat.] | T (min) | Conv. (%) | Mn (104 g/mol) | PDI (Mw/Mn) | rr (%) | mr (%) | mm (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | AMA | 5 | - | 100 | 1440 | 32 | 2.12 | 1.45 | 75.3 | 21.5 | 3.2 |
| 2 | AMA | 5 | 1 | 100 | 20 | 100 | 4.23 | 1.42 | 78.4 | 19.6 | 2.0 |
| 3 | VMA | 6 | - | 100 | 1440 | 0 | - | - | - | - | - |
| 4 | VMA | 6 | 1 | 100 | 20 | 100 | 4.89 | 1.42 | 77.4 | 21.7 | 0.9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, P.; Wu, L.; Dong, L.; Xu, X. Chemoselective Polymerization of Polar Divinyl Monomers with Rare-Earth/Phosphine Lewis Pairs. Molecules 2018, 23, 360. https://doi.org/10.3390/molecules23020360
Xu P, Wu L, Dong L, Xu X. Chemoselective Polymerization of Polar Divinyl Monomers with Rare-Earth/Phosphine Lewis Pairs. Molecules. 2018; 23(2):360. https://doi.org/10.3390/molecules23020360
Chicago/Turabian StyleXu, Pengfei, Lei Wu, Liqiu Dong, and Xin Xu. 2018. "Chemoselective Polymerization of Polar Divinyl Monomers with Rare-Earth/Phosphine Lewis Pairs" Molecules 23, no. 2: 360. https://doi.org/10.3390/molecules23020360