Design, Synthesis, and Biological Evaluation of 2-(Benzylamino-2-Hydroxyalkyl)Isoindoline-1,3-Diones Derivatives as Potential Disease-Modifying Multifunctional Anti-Alzheimer Agents

 ,
,  ,
, 
Abstract
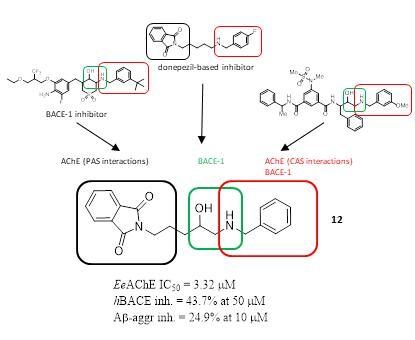
:
1. Introduction
2. Results and Discussion
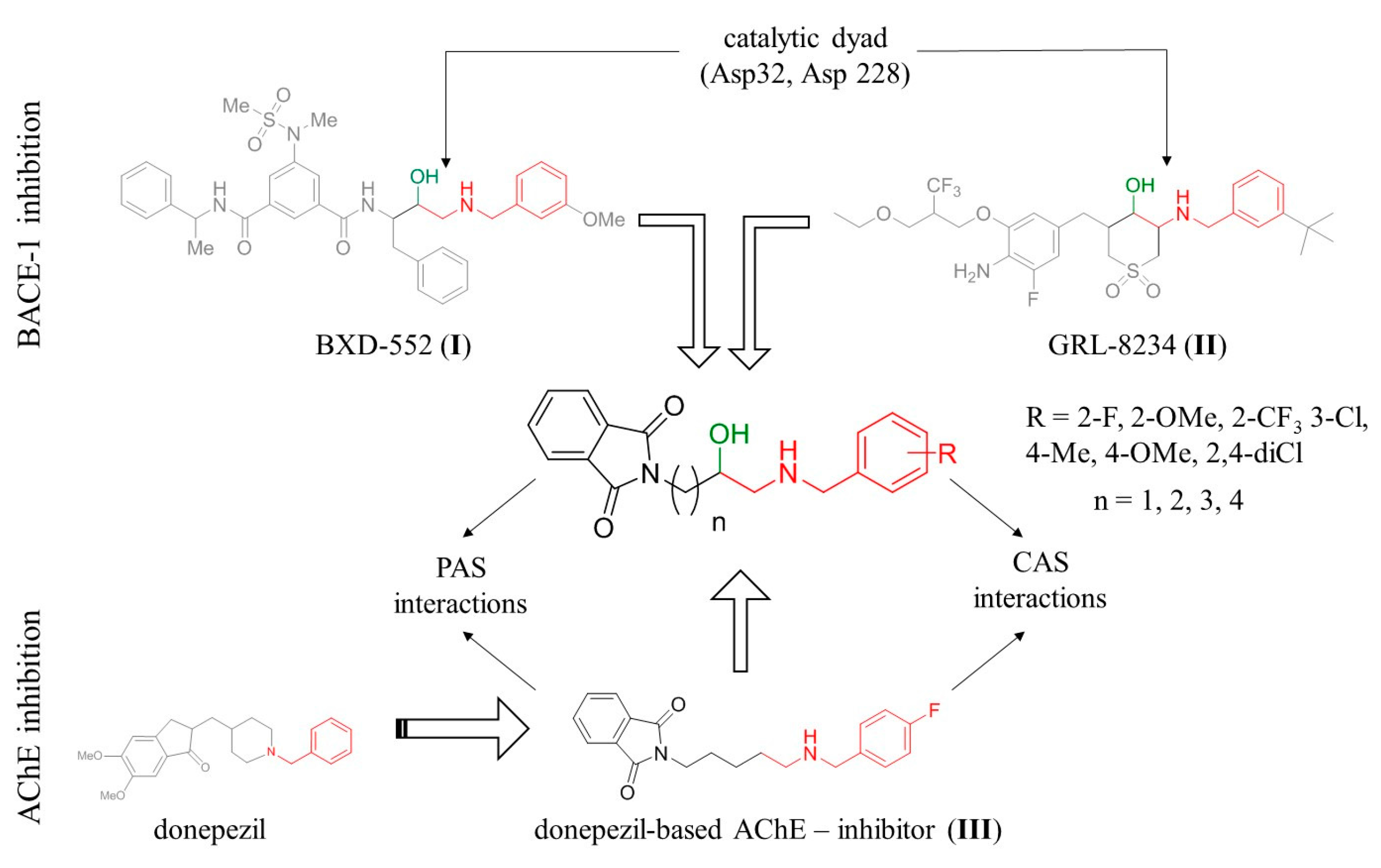
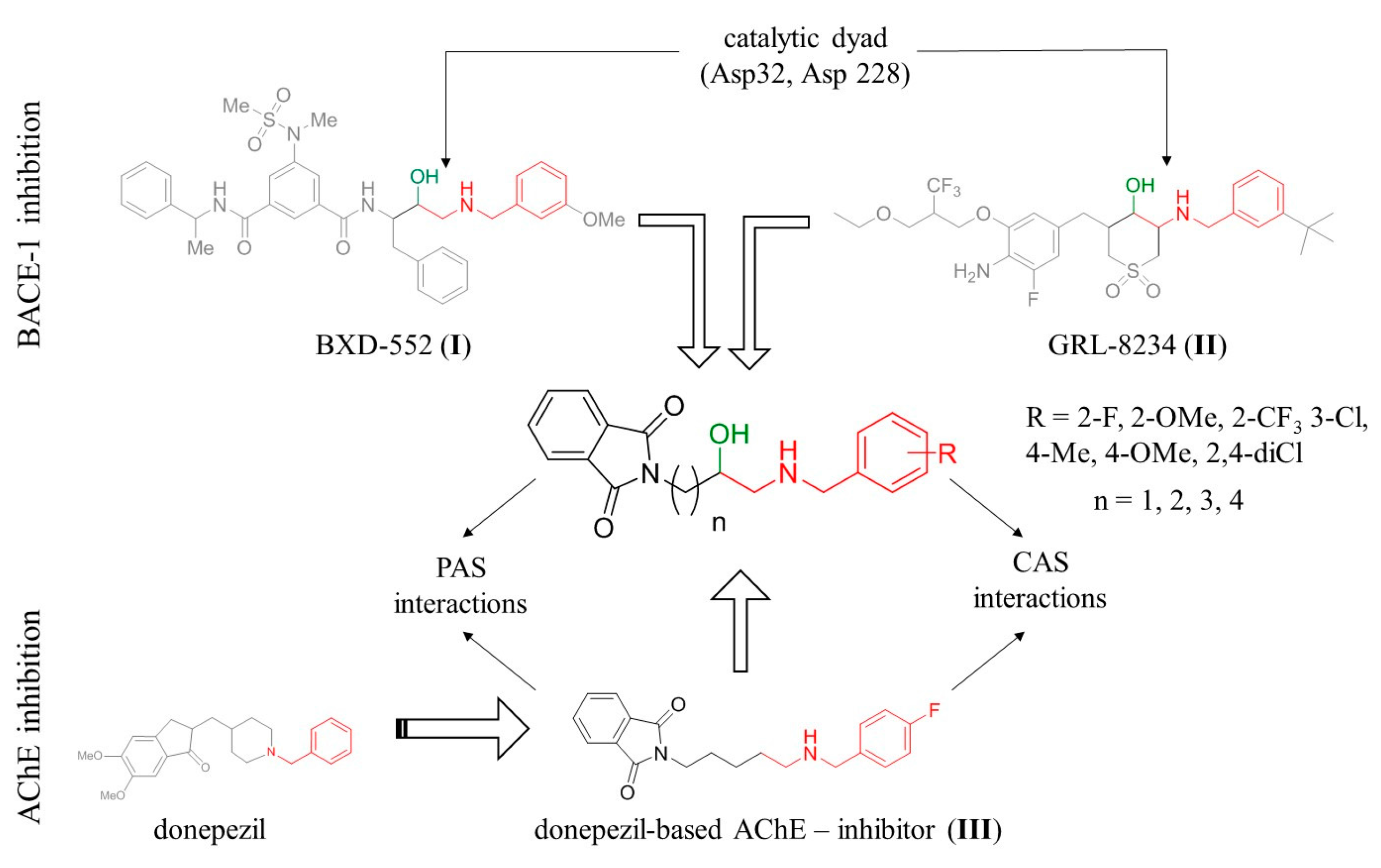
2.1. Design
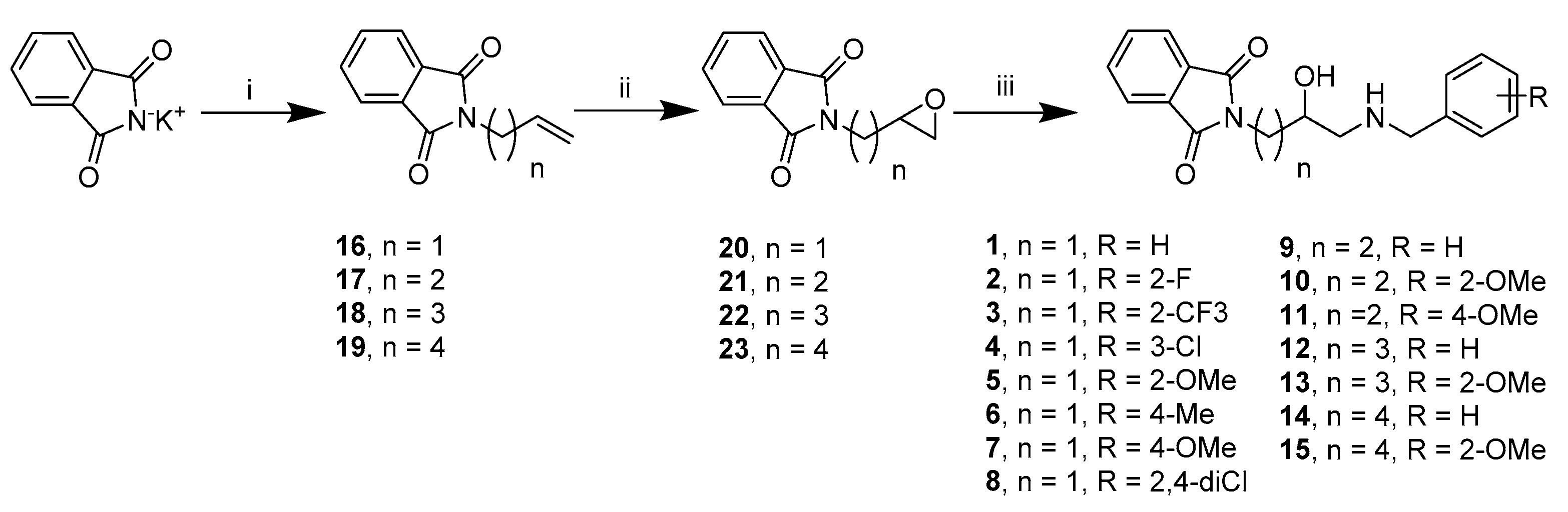
2.2. Chemistry
2.3. Biological Evaluation
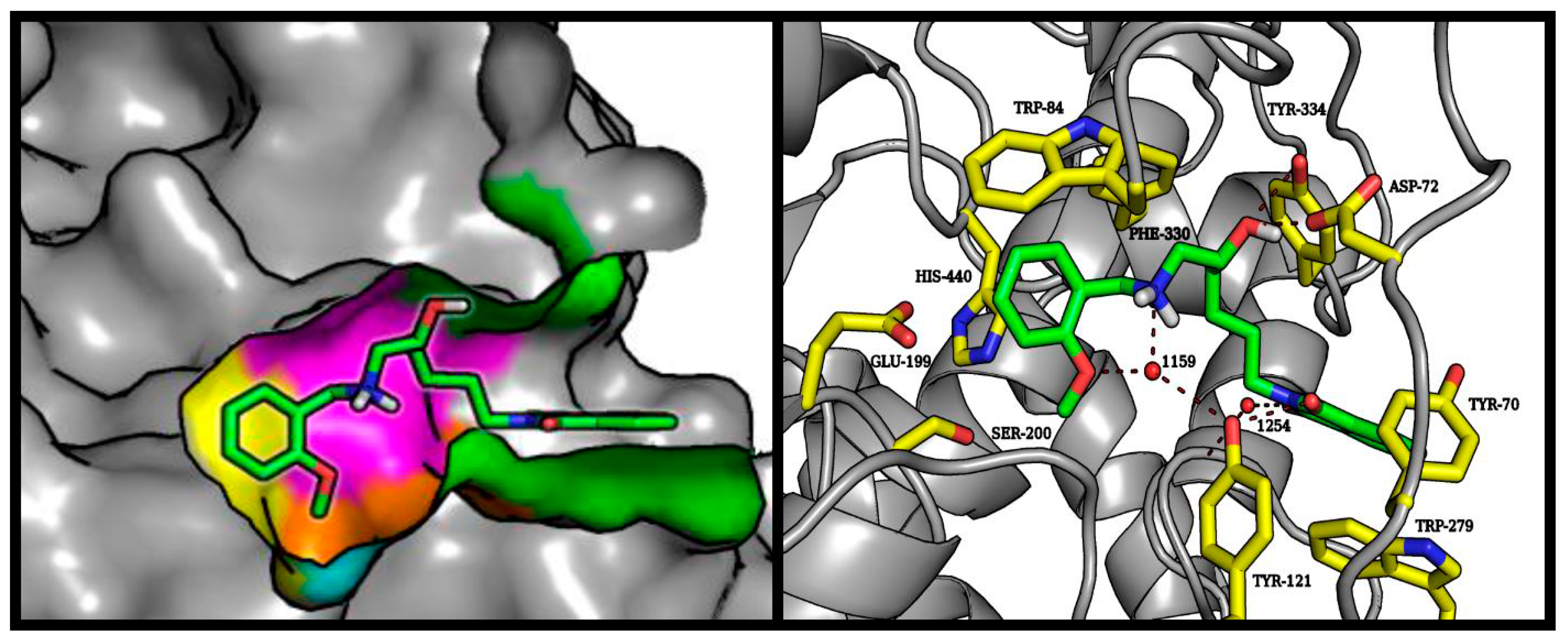
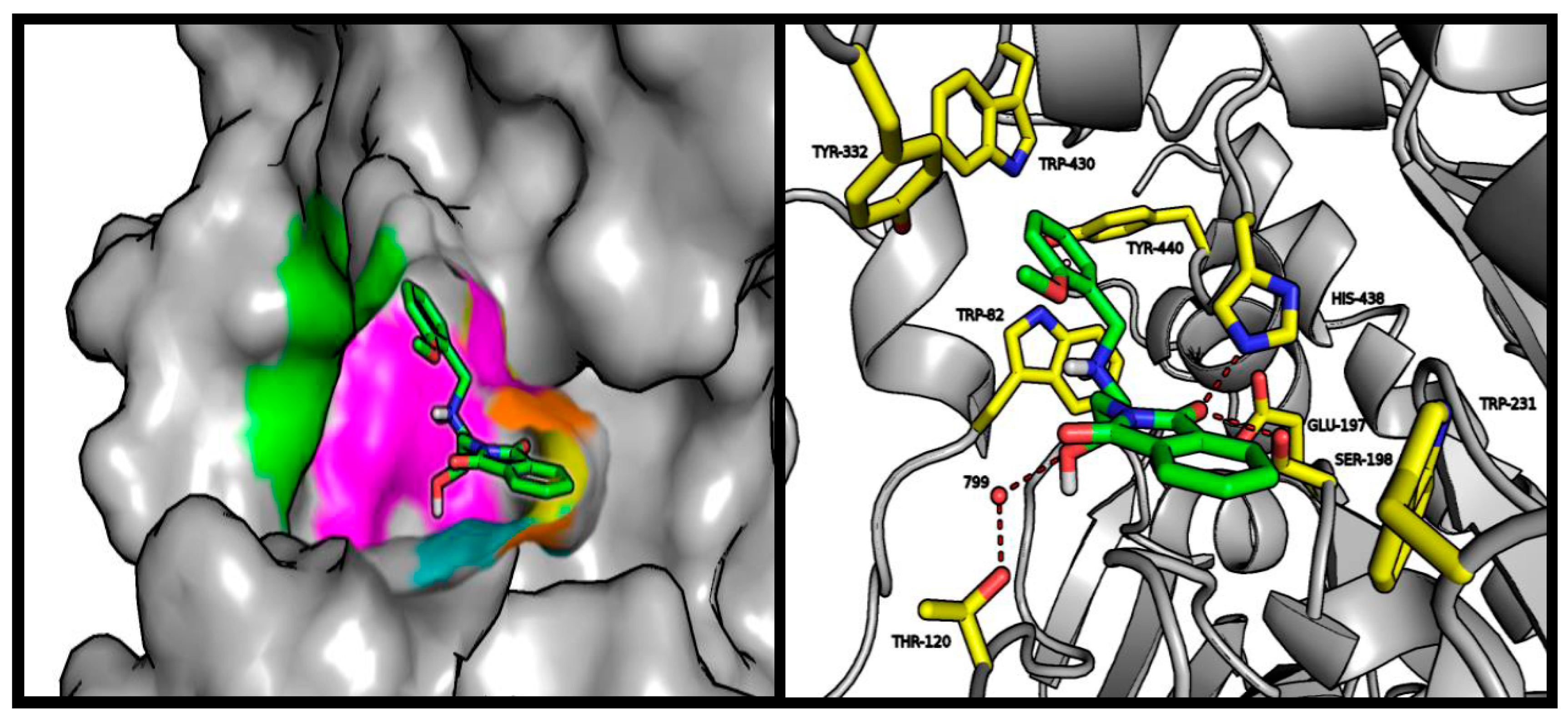
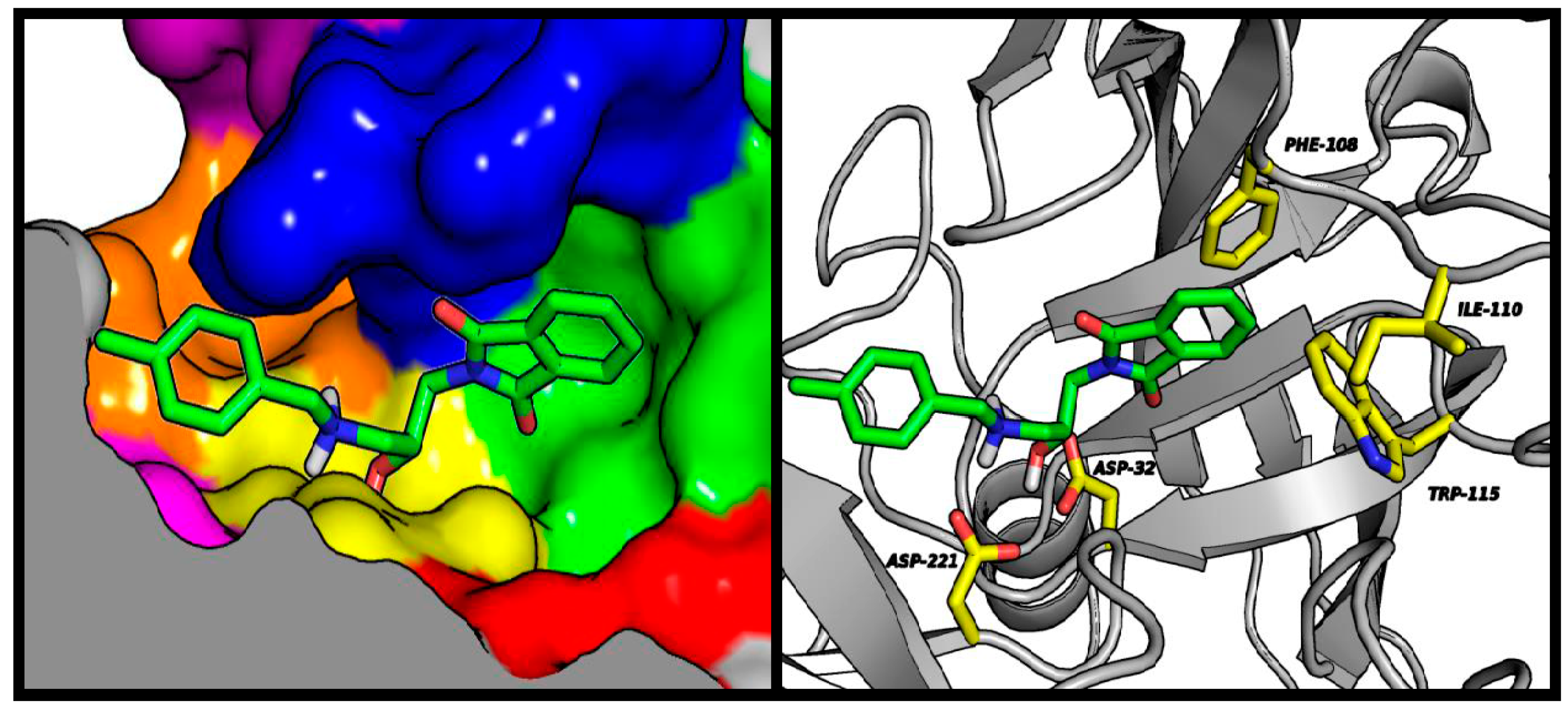
2.4. Molecular Modeling Studies
3. Materials and Methods
3.1. Chemistry
3.1.1. General Methods
3.1.2. General Procedure for the Synthesis of Compounds: 1–15 (Procedure A)
3.2. Biological Activity
3.2.1. In Vitro Inhibition of AChE and BuChE
3.2.2. In Vitro BACE-1 Inhibitory Activity
3.2.3. Inhibition of Aβ-Aggregation
3.3. Molecular Modeling
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rizzi, L.; Rosset, I.; Roriz-Cruz, M. Global Epidemiology of Dementia: Alzheimer’s and Vascular Types. Biomed. Res. Int. 2014, 2014, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Amyloid protein and Alzheimer’s disease. Sci. Am. 1991, 265, 68–71, 74–76, 78. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Pinto, T.; Lanctôt, K.L.; Herrmann, N. Revisiting the cholinergic hypothesis of behavioral and psychological symptoms in dementia of the Alzheimer’s type. Ageing Res. Rev. 2011, 10, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Bowen, D.M.; Smith, C.B.; White, P.; Davison, A.N. Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain 1976, 99, 459–496. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J.F. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 308, 1403. [Google Scholar] [CrossRef]
- Casey, D.A.; Antimisiaris, D.; O’Brien, J. Drugs for Alzheimer’s disease: Are they effective? Pharm. Ther. 2010, 35, 208–211. [Google Scholar]
- Morphy, R.; Rankovic, Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef] [PubMed]
- Rosini, M.; Simoni, E.; Caporaso, R.; Minarini, A. Multitarget strategies in Alzheimer’s disease: Benefits and challenges on the road to therapeutics. Future Med. Chem. 2016, 8, 697–711. [Google Scholar] [CrossRef] [PubMed]
- Prati, F.; Cavalli, A.; Bolognesi, M. Navigating the Chemical Space of Multitarget-Directed Ligands: From Hybrids to Fragments in Alzheimer’s Disease. Molecules 2016, 21, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spilovska, K.; Korabecny, J.; Nepovimova, E.; Dolezal, R.; Mezeiova, E.; Soukup, O.; Kuca, K. Multitarget Tacrine Hybrids with Neuroprotective Properties to Confront Alzheimer’s Disease. Curr. Top. Med. Chem. 2017, 17, 1006–1026. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, T.; Shakeri, A.; Rao, P.P.N. Amyloid cascade in Alzheimer’s disease: Recent advances in medicinal chemistry. Eur. J. Med. Chem. 2016, 113, 258–272. [Google Scholar] [CrossRef] [PubMed]
- Ismaili, L.; Refouvelet, B.; Benchekroun, M.; Brogi, S.; Brindisi, M.; Gemma, S.; Campiani, G.; Filipic, S.; Agbaba, D.; Esteban, G.; et al. Multitarget compounds bearing tacrine- and donepezil-like structural and functional motifs for the potential treatment of Alzheimer’s disease. Prog. Neurobiol. 2017, 151, 4–34. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, T.; Rao, P.P.N. 2,4-Disubstituted quinazolines as amyloid-β aggregation inhibitors with dual cholinesterase inhibition and antioxidant properties: Development and structure-activity relationship (SAR) studies. Eur. J. Med. Chem. 2017, 126, 823–843. [Google Scholar] [CrossRef] [PubMed]
- Sola, I.; Aso, E.; Frattini, D.; López-González, I.; Espargaró, A.; Sabaté, R.; Di Pietro, O.; Luque, F.J.; Clos, M.V.; Ferrer, I.; et al. Novel levetiracetam derivatives that are effective against the Alzheimer-like phenotype in mice: Synthesis, in vitro, ex vivo, and in vivo efficacy studies. J. Med. Chem. 2015, 58, 6018–6032. [Google Scholar] [CrossRef] [PubMed]
- Darras, F.H.; Pockes, S.; Huang, G.; Wehle, S.; Strasser, A.; Wittmann, H.-J.; Nimczick, M.; Sotriffer, C.A.; Decker, M. Synthesis, Biological Evaluation, and Computational Studies of Tri- and Tetracyclic Nitrogen-Bridgehead Compounds as Potent Dual-Acting AChE Inhibitors and hH3 Receptor Antagonists. ACS Chem. Neurosci. 2014, 5, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Aguilera, Ó.M.; Hagenow, S.; Palomino-Antolin, A.; Farré-Alins, V.; Ismaili, L.; Joffrin, P.-L.; Jimeno, M.L.; Soukup, O.; Janočková, J.; Kalinowsky, L.; et al. Multitarget-Directed Ligands Combining Cholinesterase and Monoamine Oxidase Inhibition with Histamine H3R Antagonism for Neurodegenerative Diseases. Angew. Chem. Int. Ed. 2017, 56, 12765–12769. [Google Scholar] [CrossRef] [PubMed]
- Guzior, N.; Bajda, M.; Skrok, M.; Kurpiewska, K.; Lewiński, K.; Brus, B.; Pišlar, A.; Kos, J.; Gobec, S.; Malawska, B. Development of multifunctional, heterodimeric isoindoline-1,3-dione derivatives as cholinesterase and β-amyloid aggregation inhibitors with neuroprotective properties. Eur. J. Med. Chem. 2015, 92, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Rueeger, H.; Rondeau, J.-M.; McCarthy, C.; Möbitz, H.; Tintelnot-Blomley, M.; Neumann, U.; Desrayaud, S. Structure based design, synthesis and SAR of cyclic hydroxyethylamine (HEA) BACE-1 inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 1942–1947. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Kumaragurubaran, N.; Hong, L.; Kulkarni, S.; Xu, X.; Miller, H.B.; Reddy, D.S.; Weerasena, V.; Turner, R.; Chang, W.; et al. Potent memapsin 2 (beta-secretase) inhibitors: Design, synthesis, protein-ligand X-ray structure, and in vivo evaluation. Bioorg. Med. Chem. Lett. 2008, 18, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Kennedy, M.E.; Wang, W.; Song, L.; Lee, J.; Zhang, L.; Wong, G.; Wang, L.; Parker, E. Measuring human β-secretase (BACE1) activity using homogeneous time-resolved fluorescence. Anal. Biochem. 2003, 319, 49–55. [Google Scholar] [CrossRef]
- Mancini, F.; De Simone, A.; Andrisano, V. Beta-secretase as a target for Alzheimer’s disease drug discovery: An overview of in vitro methods for characterization of inhibitors. Anal. Bioanal. Chem. 2011, 400, 1979–1996. [Google Scholar] [CrossRef] [PubMed]
- Stachel, S.J.; Coburn, C.A.; Steele, T.G.; Jones, K.G.; Loutzenhiser, E.F.; Gregro, A.R.; Rajapakse, H.A.; Lai, M.-T.; Crouthamel, M.-C.; Xu, M.; et al. Structure-based design of potent and selective cell-permeable inhibitors of human beta-secretase (BACE-1). J. Med. Chem. 2004, 47, 6447–6450. [Google Scholar] [CrossRef] [PubMed]
- LeVine, H. Thioflavine T interaction with synthetic Alzheimer’s disease beta-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci. 1993, 2, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Bajda, M.; Jończyk, J.; Malawska, B.; Filipek, S. Application of computational methods for the design of BACE-1 inhibitors: Validation of in silico modelling. Int. J. Mol. Sci. 2014, 15, 5128–5139. [Google Scholar] [CrossRef] [PubMed]
- Bajda, M.; Więckowska, A.; Hebda, M.; Guzior, N.; Sotriffer, C.A.; Malawska, B. Structure-based search for new inhibitors of cholinesterases. Int. J. Mol. Sci. 2013, 14, 5608–5632. [Google Scholar] [CrossRef] [PubMed]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal Structure of Human Butyrylcholinesterase and of Its Complexes with Substrate and Products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [PubMed]
- Mosley, C.A.; Myers, S.J.; Murray, E.E.; Santangelo, R.; Tahirovic, Y.A.; Kurtkaya, N.; Mullasseril, P.; Yuan, H.; Lyuboslavsky, P.; Le, P.; et al. Synthesis, structural activity-relationships, and biological evaluation of novel amide-based allosteric binding site antagonists in NR1A/NR2B N-methyl-d-aspartate receptors. Bioorg. Med. Chem. 2009, 17, 6463–6480. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, N.Y.; Tikhov, R.M.; Godovikov, I.A.; Khrustalev, V.N.; Bubnov, Y.N. New enolate-carbodiimide rearrangement in the concise synthesis of 6-amino-2,3-dihydro-4-pyridinones from homoallylamines. Org. Biomol. Chem. 2016, 14, 4283–4298. [Google Scholar] [CrossRef] [PubMed]
- Nickels, M.; Xie, J.; Cobb, J.; Gore, J.C.; Pham, W. Functionalization of iron oxide nanoparticles with a versatile epoxy amine linker. J. Mater. Chem. 2010, 20, 4776–4780. [Google Scholar] [CrossRef] [PubMed]
- Fraunhoffer, K.J.; Bachovchin, D.A.; White, M.C. Hydrocarbon Oxidation vs C−C Bond-Forming Approaches for Efficient Syntheses of Oxygenated Molecules. Org. Lett. 2005, 7, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Kanoh, S.; Naka, M.; Nishimura, T.; Motoi, M. Isomerization of cyclic ethers having a carbonyl functional group: New entries into different heterocyclic compounds. Tetrahedron 2002, 58, 7094–7096. [Google Scholar] [CrossRef]
- Dey, S.; Powell, D.R.; Hu, C.; Berkowitz, D.B. “Cassette” In Situ Enzymatic Screening Identifies Complementary Chiral Scaffolds for Hydrolytic Kinetic Resolution Across a Range of Epoxides. Angew. Chem. Int. Ed. 2007, 46, 7010–7014. [Google Scholar] [CrossRef] [PubMed]
- CombiGlide, version 4.1; Schrödinger LLC: New York, NY, USA, 2016.
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- PyMOL, version 0.99rc6; DeLano Scientific LLC: Palo Alto, CA, USA, 2006.
Sample Availability: Samples of the selected compounds (1–15) are available from the authors. |
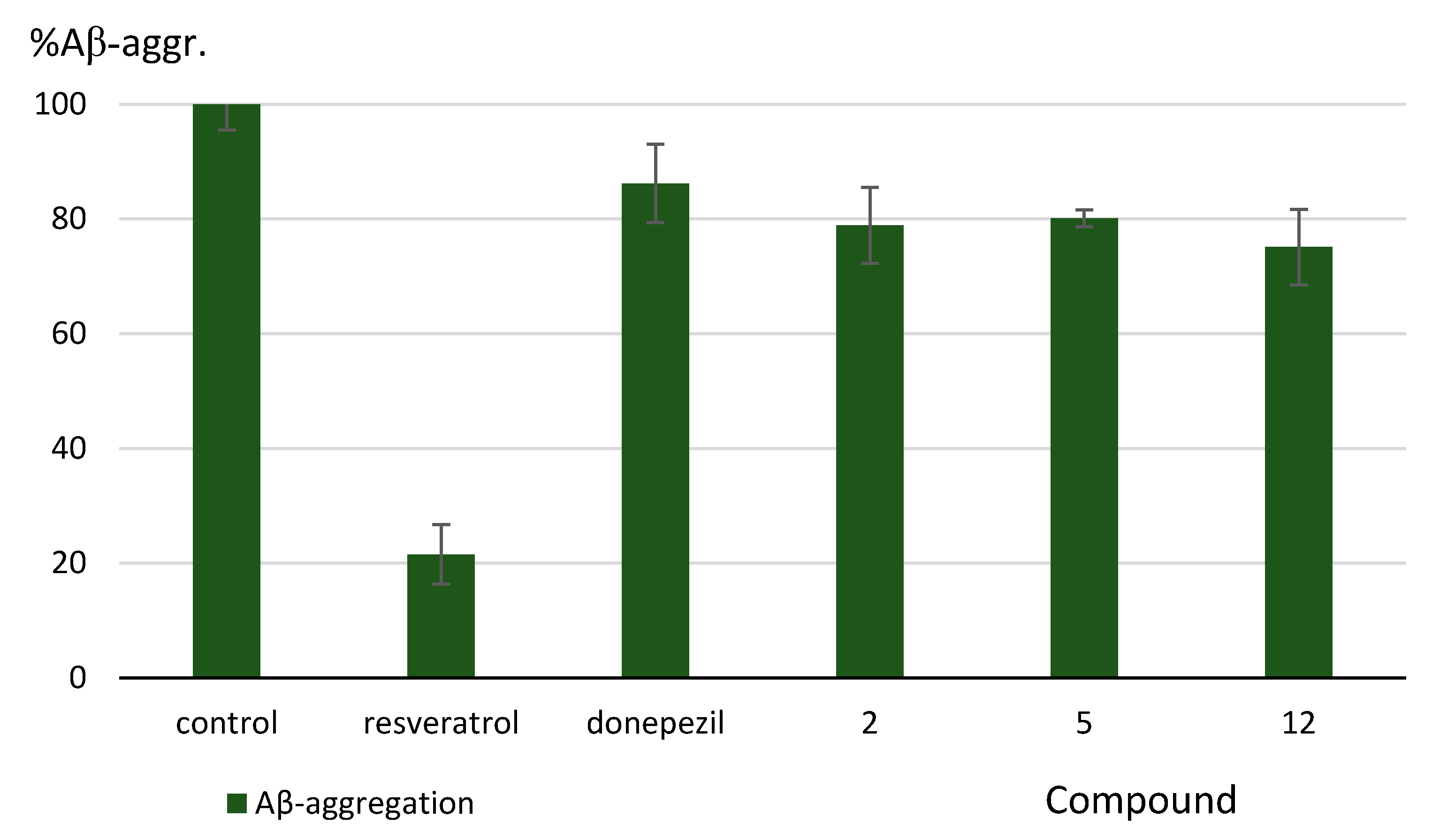

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
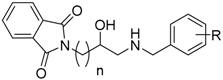 | ||||||
|---|---|---|---|---|---|---|
| Cmp. | R | n | eeAChE a | eqBuChE b | hBACE-1 c. | Aβ-Aggr. d |
| IC50 (µM) e/% Inh. f | IC50 (µM) e/% Inh. f | % Inh. f | % Inh. f | |||
| 1 | H | 1 | 11.5% ± 5.0% | 14.1% ± 3.3% | 37.3% ± 7.5% | <10% |
| 2 | 2-F | 1 | 28.1% ± 2.0% | 19.5% ± 2.0% | 39.6% ± 6.5% | 21.1% ± 6.6% |
| 3 | 2-CF3 | 1 | 11.8% ± 5.0% | <10% | 41.8% ± 4.5% | <10% |
| 4 | 3-Cl | 1 | <10% | 24.1% ± 4.7% | 34.1% ± 7.7% | <10% |
| 5 | 2-OMe | 1 | <10% | 7.86 ± 0.29 | 39.2% ± 9.2% | 19.9% ± 1.5% |
| 6 | 4-Me | 1 | <10% | 11.6% ± 3.3% | 45.0% ± 2.0% | <10% |
| 7 | 4-OMe | 1 | <10% | 19.1% ± 1.5% | 35.1% ± 10.0% | <10% |
| 8 | 2,4-diCl | 1 | <10% | 21.7% ± 1.0% | 37.2% ± 1.3% | <10% |
| 9 | H | 2 | <10% | <10% | <10% | <10% |
| 10 | 2-OMe | 2 | 32.7% ± 4.7% | 17.8% ± 2.0% | 24.4% ± 5.9% | <10% |
| 11 | 4-OMe | 2 | <10% | <10% | 34.8% ± 10.6% | <10% |
| 12 | H | 3 | 3.32 ± 0.15 | 14.1% ± 5.8% | 43.7% ± 6.9% | 24.9% ± 6.6% |
| 13 | 2-OMe | 3 | 11.07 ± 0.49 | 26.2% ± 9.7% | 37.1% ± 3.9% | <10% |
| 14 | H | 4 | 2.13 ± 0.07 | 36.0% ± 7.2% | 42.1% ± 10.7% | <10% |
| 15 | 2-OMe | 4 | 1.95 ± 0.06 | 36.0% ± 1.1% | 15.7% ± 1.3% | <10% |
| Reference compounds | ||||||
| Donepezil | 0.011 ± 0.0002 | 1.83 ± 0.04 | n.d. g | 13.8% ± 6.8 | ||
| Tacrine | 0.023 ± 0.0004 | 0.015 ± 0.0001 | n.d. g | n.d. g | ||
| Inhibitor IV h | n.d. g | n.d. g | 0.046 ± 0.001 i | n.d. g | ||
| Resveratrol | n.d. g | n.d. g | n.d. g | 78.5% ± 5.2 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panek, D.; Więckowska, A.; Pasieka, A.; Godyń, J.; Jończyk, J.; Bajda, M.; Knez, D.; Gobec, S.; Malawska, B. Design, Synthesis, and Biological Evaluation of 2-(Benzylamino-2-Hydroxyalkyl)Isoindoline-1,3-Diones Derivatives as Potential Disease-Modifying Multifunctional Anti-Alzheimer Agents. Molecules 2018, 23, 347. https://doi.org/10.3390/molecules23020347
Panek D, Więckowska A, Pasieka A, Godyń J, Jończyk J, Bajda M, Knez D, Gobec S, Malawska B. Design, Synthesis, and Biological Evaluation of 2-(Benzylamino-2-Hydroxyalkyl)Isoindoline-1,3-Diones Derivatives as Potential Disease-Modifying Multifunctional Anti-Alzheimer Agents. Molecules. 2018; 23(2):347. https://doi.org/10.3390/molecules23020347
Chicago/Turabian StylePanek, Dawid, Anna Więckowska, Anna Pasieka, Justyna Godyń, Jakub Jończyk, Marek Bajda, Damijan Knez, Stanislav Gobec, and Barbara Malawska. 2018. "Design, Synthesis, and Biological Evaluation of 2-(Benzylamino-2-Hydroxyalkyl)Isoindoline-1,3-Diones Derivatives as Potential Disease-Modifying Multifunctional Anti-Alzheimer Agents" Molecules 23, no. 2: 347. https://doi.org/10.3390/molecules23020347