Chromatographic and Spectroscopic Identification and Recognition of Natural Dyes, Uncommon Dyestuff Components, and Mordants: Case Study of a 16th Century Carpet with Chintamani Motifs

 ,
,
Abstract

1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
2.2. Origin of Textile Fibre Samples
2.3. Extraction of Dyes from Threads
2.4. Equipment
3. Results and Discussion
3.1. Microscopic and Spectroscopic Studies
3.1.1. Surface Morphology
3.1.2. Fourier Transformation Infrared Spectroscopy Analysis
3.1.3. Scanning Electron Microscopy with Energy-Dispersive X-ray Detector Analysis
3.2. High-Performance Liquid Chromatography-Mass Spectrometry Analysis
3.2.1. Yellow Fibres
3.2.2. Red Fibres
3.2.3. Blue and Green Fibres
3.2.4. Beige and Brown Fibres
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pozzi, F.; Zaleski, S.; Casadio, F.; Leona, M.; Lombardi, J.R.; Van Duyne, R.P. Surface-enhanced Raman spectroscopy: Using nanoparticles to detect trace amounts of colorants in works of art. In Nanoscience and Cultural Heritage; Dillmann, P., Bellot-Gurlet, L., Nenner, I., Eds.; Atlantis Press: Amsterdam, The Netherlands, 2016; pp. 161–204. ISBN 9789462391970. [Google Scholar]
- Sultan, S.; Kareem, K.; He, L.; Simon, S. Identification of the authenticity of pigments in ancient polychromed artworks of China. Anal. Methods 2015, 9, 814–825. [Google Scholar] [CrossRef]
- Degano, I.; Biesaga, M.; Colombini, M.P.; Trojanowicz, M. Historical and archaeological textiles: An insight on degradation products of wool and silk yarns. J. Chromatogr. A 2011, 1218, 5837–5847. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.; Fisher, A.; Gibson, B.; Marshall, J.; Russelle, B.; Whitesidef, I. Atomic spectrometry update: Review of advances in the analysis of metals, chemicals and materials. J. Anal. At. Spectrom. 2017, 32, 2068–2117. [Google Scholar] [CrossRef]
- Madariaga, J.M. Analytical chemistry in the field of cultural heritage. Anal. Methods 2015, 7, 4848–4876. [Google Scholar] [CrossRef]
- Cardell, C.; Guerra, I. An overview of emerging hyphenated SEM-EDX and Raman spectroscopy systems: Applications in life, environmental and materials sciences. Trends Anal. Chem. 2016, 77, 156–166. [Google Scholar] [CrossRef]
- Wouters, J. High performance liquid chromatography of anthraquinones: Analysis of plant and insect extracts and dyed textiles. Stud. Conserv. 1985, 30, 119–128. [Google Scholar] [CrossRef]
- Halpine, S.M. An improved dye and lake pigment analysis method for high-performance liquid chromatography and diode-array detector. Stud. Conserv. 1996, 41, 76–94. [Google Scholar] [CrossRef]
- Pauk, V.; Bartak, P.; Lemr, K. Characterization of natural organic colorants in historical and art objects by high-performance liquid chromatography. J. Sep. Sci. 2014, 37, 3393–3410. [Google Scholar] [CrossRef] [PubMed]
- Walton, P.; Taylor, G. The characterization of dyes in textiles from archaeological excavations. Chromatogr. Anal. 1991, 17, 5–7. [Google Scholar]
- Taylor, G.W. Detection and identification of dyes on Anglo-Scandinavian textiles. Stud. Conserv. 1983, 28, 153–160. [Google Scholar] [CrossRef]
- Benkendorff, K.; Bremner, J.; Davis, A. Indole Derivatives from the Egg Masses of Muricid Molluscs. Molecules 2001, 6, 70–78. [Google Scholar] [CrossRef]
- Pawlak, K.; Puchalska, M.; Miszczak, A.; Rosłoniec, E.; Jarosz, M. Blue natural organic dyestuffs—From textile dyeing to mural painting. Separation and characterization of coloring matters present in elderberry, logwood and indigo. J. Mass Spectrom. 2006, 41, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Mantzouris, D.; Karapanagiotis, I.; Panayiotou, C. Comparison of extraction methods for the analysis of Indigofera tinctoria and Carthamus tinctorius in textiles by high performance. Microchem. J. 2014, 115, 78–86. [Google Scholar] [CrossRef]
- Otłowska, O.; Ślebioda, M.; Wachowiak, M.; Śliwka-Kaszyńska, M. Identification and characterization of the Indian Yellow dyestuff and its degradation products in historical oil paint tube by liquid chromatography mass spectrometry. RSC Adv. 2015, 5, 48786–48792. [Google Scholar] [CrossRef]
- Sanyova, J.; Reisse, J. Development of a mild method for the extraction of anthraquinones from their aluminum complexes in madder lakes prior to HPLC analysis. J. Cult. Herit. 2006, 7, 229–235. [Google Scholar] [CrossRef]
- Otłowska, O.; Ślebioda, M.; Wachowiak, M.; Śliwka-Kaszyńska, M. A multi-analytical approach to the characterization of natural organic dyestuffs and inorganic substrates present in the 19th-century artistic oil paints manufactured by a French art materials supplier Richard Aines. Anal. Methods 2017, 9, 94–102. [Google Scholar] [CrossRef]
- Valianou, L.; Karapanagiotis, I.; Chryssoulakis, Y. Comparison of extraction methods for the analysis of natural dyes in historical textiles by high-performance liquid chromatography. Anal. Bioanal. Chem. 2009, 395, 2175–2189. [Google Scholar] [CrossRef] [PubMed]
- Ticha, M.B.; Meksi, N.; Attia, H.E.; Haddar, W.; Guesmi, A.; Jannet, H.B.; Mhenni, M.F. Ultrasonic extraction of Parthenocissus quinquefolia colorants: Extract identification by HPLC-MS analysis and cleaner application on the phytodyeing of natural fibres. Dyes Pigment 2017, 141, 103–111. [Google Scholar] [CrossRef]
- Degano, I.; Tognotti, P.; Kunzelman, D.; Modugno, F. HPLC-DAD and HPLC-ESI-Q-ToF characterisation of early 20th century lake and organic pigments from Lefranc archives. Herit. Sci. 2017. [Google Scholar] [CrossRef]
- Papliaka, Z.E.; Konstanta, A.; Karapanagiotis, I.; Karadag, R.; Akyol, A.A.; Mantzouris, D.; Tsiamyrtzis, P. FTIR imaging and HPLC reveal ancient painting and dyeing techniques of molluskan purple. Archaeol. Anthropol. Sci. 2017, 9, 197–208. [Google Scholar] [CrossRef]
- Kramell, A.E.; Wertmann, P.; Hosner, D.; Kluge, R.; Oehler, F.; Wunderlich, C.H.; Tarasov, P.E.; Wagner, M.; Csuk, R. A multi-analytical techniques based approach to study the colorful clothes and accessories from mummies of Eastern Central Asia. J. Archaeol. Sci. Rep. 2016, 10, 464–473. [Google Scholar] [CrossRef]
- Peets, P.; Leitoa, I.; Pelt, J.; Vahur, S. Identification and classification of textile fibres using ATR-FT-IR spectroscopy with chemometric methods. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 173, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Akyuz, S.; Akyuz, T.; Cakan, B.; Basaran, S. Investigations of the historic textiles excavated from Ancient Ainos (Enez-Turkey) by multiple analytical techniques. J. Mol. Struct. 2014, 1073, 37–43. [Google Scholar] [CrossRef]
- Kakkar, P.; Madhan, B.; Shanmugamet, G. Extraction and characterization of keratin from bovine hoof: A potential material for biomedical applications. SpringerPlus 2014, 3, 596. [Google Scholar] [CrossRef] [PubMed]
- Joosten, I.; van Bommel, M.R.; Hofmann de Keijzer, R.; Reschreiter, H. Micro Analysis on Hallstatt Textiles: Colour and Condition. Microchim. Acta 2006, 155, 169–174. [Google Scholar] [CrossRef]
- Hofenk de Graaff, J.H. The colourful past. In Origins Chemistry and Identification of Natural Dyestuffs; Abegg Stiftung and Archetype Publications Ltd.: London, UK, 2004; ISBN 1873132131. [Google Scholar]
- Schweppe, H. Handbuch der Naturfarbstoffe. Vorkommen—Verwendung—Nachweis; Ecomed VIg: Landsberg, Germany, 1993; ISBN 978-3933203465. [Google Scholar]
- Stobiecki, M.; Kachlicki, P.; Wojakowska, A.; Marczak, Ł. Application of LC/MS systems to structural characterization of flavonoid glycoconjugates. Phytochem. Lett. 2015, 11, 358–367. [Google Scholar] [CrossRef]
- Quin, Y.; Gao, B.; Shi, H.; Cao, J.; Yin, C.; Lu, W.; Yu, L.; Cheng, Z. Characterization of flavonol mono-, di-, tri- and tetra-O-glycosides by ultra-performance liquid chromatography-electrospray ionization-quadrupole time-of-flight mass spectrometry and its application for identification of flavonol glycosides in Viola tianschanica. J. Pharm. Biomed. Anal. 2017, 142, 113–124. [Google Scholar] [CrossRef]
- Moiteiro, C.; Gaspar, H.; Rodrigues, A.I.; Lopes, J.F.; Carnide, V. HPLC quantification of dye flavonoids in Reseda luteola L. from Portugal. J. Sep. Sci. 2008, 31, 3683–3687. [Google Scholar] [CrossRef] [PubMed]
- Marques, R.; Sousa, M.M.; Oliveira, M.C.; Melo, M.J. Characterization of weld (Reseda luteola L.) and spurge flax (Daphne gnidium L.) by high-performance liquid chromatography–diode array detection–mass spectrometry in Arraiolos historical textiles. J. Chromatogr. A 2009, 1216, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- March, R.E.; Levars, E.G.; Stadey, C.J.; Miao, X.S.; Zhao, X.; Metcalfe, C.D. A comparison of flavonoid glycosides by electrospray tandem mass spectrometry. Int. J. Mass Spectrom. 2006, 248, 61–85. [Google Scholar] [CrossRef]
- Fabre, N.; Rustan, I.; de Hoffmann, E.; Quetin-Leclercq, J. Determination of flavone, flavonol, and flavanone aglycones by negative ion liquid chromatography electrospray ion trap mass spectrometry. J. Am. Soc. Mass Spectrom. 2001, 12, 707–715. [Google Scholar] [CrossRef]
- Troalen, L.G.; Phillips, A.S.; Peggie, D.A.; Barran, P.E.; Hulme, A.N. Historical textile dyeing with Genista tinctoria L.: A comprehensive study by UPLC-MS/MS analysis. Anal. Methods 2014, 6, 8915–8923. [Google Scholar] [CrossRef]
- Valianou, L.; Stathopoulou, K.; Karapanagiotis, I.; Magiatis, P.; Pavlidou, E.; Skaltsounis, A.L.; Chryssoulakis, Y. Phytochemical analysis of young fustic (Cotinus coggygria heartwood) and identification of isolated colourants in historical textiles. Anal. Bioanal. Chem. 2009, 394, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Liu, D.; Nikolic, D.; Zhu, D.; Pezzuto, J.M.; van Breemen, R.B. In vitro metabolism of isoliquiritigenin by human liver microsomes. Drug Metab. Dispos. 2008, 36, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.J.; Kim, I.S.; Rehman, S.U.; Dong, M.S.; Na, C.S.; Yoo, H.H. A Liquid Chromatography–Tandem Mass Spectrometry Method for Simultaneous Quantitation of 10 Bioactive Components in Rhus verniciflua Extracts. J. Chromatogr. Sci. 2016, 54, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Karapanagiotis, I.; Lakka, A.; Valianou, L.; Chryssoulakis, Y. High-performance liquid chromatographic determination of colouring matters in historical garments from the Holy Mountain of Athos. Microchim. Acta 2008, 160, 477–483. [Google Scholar] [CrossRef]
- Karapanagiotis, I.; Minopoulou, E.; Valianou, L.; Daniilia, S.; Chryssoulakis, Y. Investigation of the colourants used in icons of the Cretan School of iconography. Anal. Chim. Acta 2009, 647, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Hulme, A.N.; McNab, H.; Peggie, D.A.; Quye, A. Negative ion electrospray mass spectrometry of neoflavonoids. Phytochemistry 2005, 66, 2766–2770. [Google Scholar] [CrossRef] [PubMed]
- Manhita, A.; Balcaend, L.; Vanhaecked, F.; Ferreira, T.; Candeiasa, A.; Dias, C.B. Unveiling the colour palette of Arraiolos carpets: Material study of carpets from the 17th to 19th century period by HPLC-DAD-MS and ICP-MS. J. Cult. Herit. 2014, 15, 292–299. [Google Scholar] [CrossRef]
- Santos, R.; Hallett, J.; Conceicao Oliveira, M.; Sousa, M.M.; Sarraguça, J.; Simmonds, M.S.J.; Nesbitt, M. HPLC-DAD-MS analysis of colorant and resinous components of lac-dye: A comparison between Kerria and Paratachardina genera. Dyes Pigment 2015, 118, 129–136. [Google Scholar] [CrossRef]
- Petroviciu, I.; Albu, F. Medvedovici, A. LC/MS and LC/MS/MS based protocol for identification of dyes in historic textiles. Microchem. J. 2010, 95, 247–254. [Google Scholar] [CrossRef]
- Rosenberg, E. Characterisation of historical organic dyestuffs by liquid chromatography-mass spectrometry. Anal. Bioanal. Chem. 2008, 391, 33–57. [Google Scholar] [CrossRef] [PubMed]
- Szostek, B.; Orska-Gawrys, J.; Surowiec, I.; Trojanowicz, M. Investigation of natural dyes occurring in historical Coptic textiles by high-performance liquid chromatography with UV-Vis and mass spectrometric detection. J. Chromatogr. A 2003, 1012, 179–192. [Google Scholar] [CrossRef]
- Ferreira, E.S.B.; Hulme, A.M.; McNaby, H.; Quye, A. The natural constituents of historical textile dyes. Chem. Soc. Rev. 2004, 33, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Petroviciu, I.; Vanden Berghe, I.; Cretu, I.; Albu, F.; Medvedovici, A. Identification of natural dyes in historical textiles from Romanian collections by LC-DAD and LC-MS (single stage and tandem MS). J. Cult. Herit. 2012, 13, 89–97. [Google Scholar] [CrossRef]
Sample Availability: not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fibre No. | F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 | F10 | F11 | F12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Element | ||||||||||||
| C | 70 | 64 | 76 | 67 | 72 | 66 | 71 | 64 | 62 | 65 | 58 | 69 |
| O | 27 | 33 | 22 | 31 | 27 | 32 | 27 | 34 | 36 | 33 | 33 | 26 |
| Al | 0.5 | 0.6 | 0.2 | 0.1 | 0.1 | 0.3 | 0.1 | 0.3 | 0.3 | 0.3 | 0.6 | 0.5 |
| Si | 0.2 | 0.3 | - | 0.1 | - | 0.1 | 0.1 | 0.4 | 0.4 | 0.1 | 0.9 | 0.6 |
| S | 0.8 | 0.5 | 1.0 | 0.7 | 0.7 | 0.7 | 0.6 | 0.5 | 0.6 | 0.5 | 3.5 | 2.7 |
| Ca | 0.5 | 0.5 | 0.4 | 0.4 | 1.0 | 0.4 | 0.5 | 0.3 | 0.5 | 0.4 | 2.1 | 0.6 |
| Fe | <0.1 | <0.1 | - | 0.15 | 0.2 | 0.1 | <0.1 | <0.1 | <0.1 | <0.1 | 1.6 | 0.1 |
| Mg | 0.2 | 0.2 | 0.2 | 0.1 | <0.1 | <0.1 | 0.1 | <0.1 | 0.1 | <0.1 | 0.2 | 0.3 |
| Na | - | 0.5 | - | - | - | - | - | - | - | - | - | 0.5 |
| Traces b | P, Cu | P | Cu | Cu | Cu | Cu | - | Cu | P, Cu | P | - | Cu |
| Peak No. | tR (min) | [M−H]−, m/z | MS2 Product Ions (m/z) | Elemental Composition | Diff (ppm) | Proposed Identification | χmax (nm) | |
|---|---|---|---|---|---|---|---|---|
| Nominal | Highly Resolved | |||||||
| Y1 | 9.6 | 593 | 593.1513 | 503, 575, 473, 383 | C27H30O15 | −0.17 | apigenin-C-diglucoside | 272, 335 |
| Y2 | 9.9 | 609 | 609.1442 | 447, 285 | C27H30O16 | 3.12 | luteolin-O-diglucoside | 268, 336 |
| Y3 | 10.6 | 609 | 609.1462 | 447, 285 | C27H30O16 | −0.16 | luteolin-3,7′-O-diglucoside | 268, 341 |
| Y4 | 11.3 | 447 | 447.0923 | 285, 284 | C21H20O11 | 2.24 | luteolin-7-O-glucoside | 268, 349 |
| Y5 | 12.2 | 447 | 447.0932 | 285 | C21H20O11 | 0.24 | luteolin-O-glucoside | 268, 337 |
| Y6 | 12.2 | 431 | 431,0989 | 311, 269, 268 | C21H20O10 | −1.16 | apigenin-7-O-glucoside | 266, 348 |
| Y7 | 12.4 | 461 | 461,1072 | 341, 299, 284, 283 * | C22H22O11 | 3.68 | chryoseriol-O-glucoside | 266, 348 |
| Y8 | 12.8 | 447 | 447.0936 | 285 | C21H20O11 | −0.67 | luteolin-4′-O-glucoside | 268, 342 |
| Y9 | 14.2 | 285 | 285.0407 | 257, 217, 199, 175, 151, 133 | C15H10O6 | −1.05 | luteolin | 255, 349 |
| Y10 | 15.3 | 269 | 269.0454 | 225, 151, 117 | C15H10O5 | 0.37 | apigenin | 267, 337 |
| Y11 | 15.5 | 299 | 299.0563 | 284, 256 | C16H12O6 | −0.67 | chryoseriol | 266, 347 |
| Y12 | 17.4 | 313 | 313.0349 | 285, 243, 201, 179, 133 | C16H10O7 | 1.60 | luteolin derivative | 248, 346 |
| Y13 | 18.0 | 313 | 313.0342 | 285, 243, 201, 179, 133 | C16H10O7 | 3.83 | luteolin derivative | 242, 346 |
| Y14 | 9.4 | 349 | 349.0028 | 371, 338, 269, 225, 213, 177, 165, 149, 135, 121 | - | - | unknown | 261, 392 |
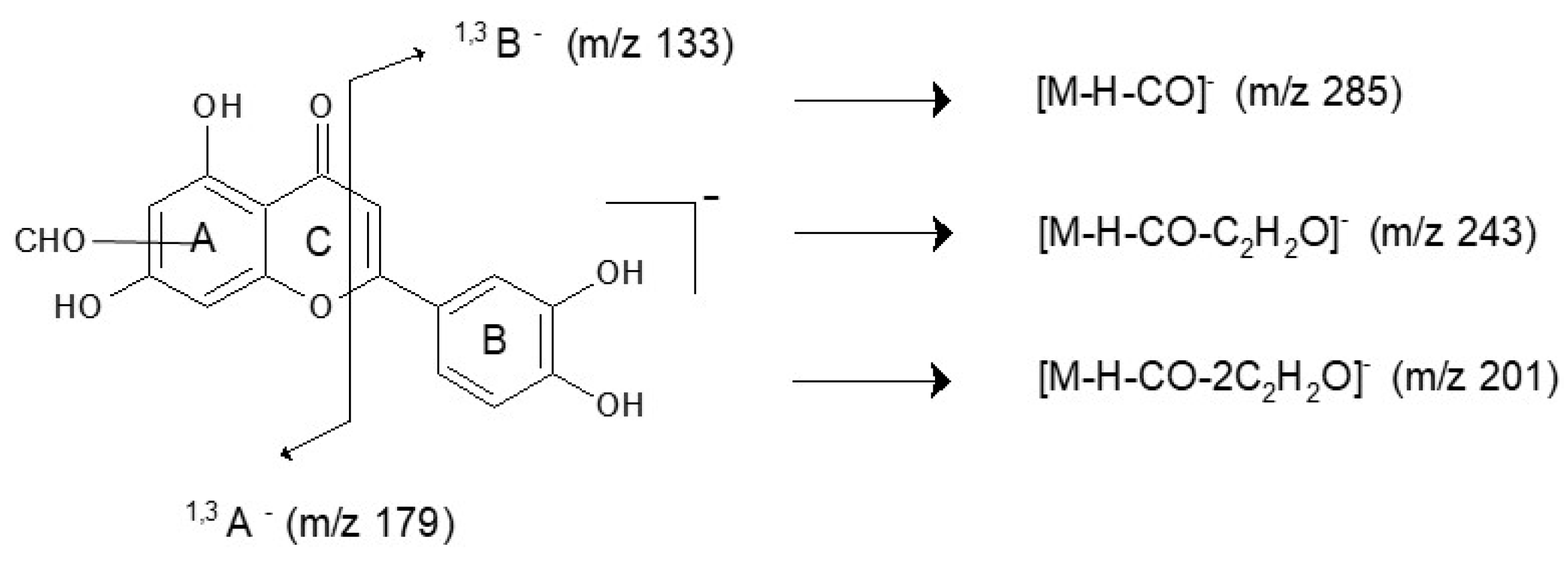
| Y15 | 12.0 | 243 | 243.0294 | 215, 199, 187, 175, 145 * | - | - | type C | 308, 336 |
| Y16 | 12.4 | 269 | 269.0448 | 241, 225,185, 135, 133 | C15H10O5 | 2.60 | sulfuretin isomer | 316, 343 |
| Y17 | 12.7 | 285 | 285.0407 | 241, 229, 149, 135, 121 | C15H10O6 | −1.05 | fistein | 320, 360 |
| Y18 | 13.5 | 269 | 269.0459 | 241, 225, 213, 195, 135 | C15H10O5 | −1.47 | sulfuretin | 256, 396 |
| Y19 | 14.9 | 314 | 314.0302 | 267, 239, 217, 199, 163, 135 | - | - | unknown | 292, 342 |
| R1 | 8.5 | 522 | 522.0656 | 478, 434 | C25H17NO12 | 4.02 | xantholaccaic acid C | 293, 425 |
| R2 | 9.0 | 538 | 538.0626 | 520, 494,476, 450, 432 | C25H17NO13 | 0.19 | laccaic acid C | 288, 490 |
| R3 | 9.5 | 494 | 494.0723 | 476, 450, 432, 406, 388, 378 | C24H17NO11 | 1.21 | laccaic acid E | 288, 490 |
| R4 | 11.0 | 552 | 552.0769 | 534, 508, 490, 464, 446 | C26H19NO13 | 2.54 | derivative of laccaic acid A | 285, 504 |
| R5 | 11.1 | 520 | 520.0854 | 502, 476, 458, 432, 414 | C26H19NO11 | 5.96 | xantholaccaic acid A | 294, 430 |
| R6 | 11.3 | 536 | 536.0836 | 518, 492, 474, 448, 430, 420 | C26H19NO12 | −0.37 | laccaic acid A | 288, 490 |
| R7 | 11.3 | 495 | 495.0568 | 477, 451, 433,407, 389 | C24H16NO12 | 0.2 | laccaic acid B | 288,490 |
| R8 | 12.5 | 606 | 606.1184 | 562, 518 | - | - | unknown | 288, 492 |
| B1 | 18.1 | 261 | 261.0665 | 233, 217 | C16H10N2O2 | 1.53 | indigotin | 288, 620 |
| B2 | 19.2 | 261 | 261.0669 | 233, 217 | C16H10N2O2 | 0 | indirubin | 290, 550 |
| BR1 | 11.5 | 301 | 300.9992 | 284, 257, 229 | C14H8O8 | 2.3 | ellagic acid | 255, 355 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otłowska, O.; Ślebioda, M.; Kot-Wasik, A.; Karczewski, J.; Śliwka-Kaszyńska, M. Chromatographic and Spectroscopic Identification and Recognition of Natural Dyes, Uncommon Dyestuff Components, and Mordants: Case Study of a 16th Century Carpet with Chintamani Motifs. Molecules 2018, 23, 339. https://doi.org/10.3390/molecules23020339
Otłowska O, Ślebioda M, Kot-Wasik A, Karczewski J, Śliwka-Kaszyńska M. Chromatographic and Spectroscopic Identification and Recognition of Natural Dyes, Uncommon Dyestuff Components, and Mordants: Case Study of a 16th Century Carpet with Chintamani Motifs. Molecules. 2018; 23(2):339. https://doi.org/10.3390/molecules23020339
Chicago/Turabian StyleOtłowska, Olga, Marek Ślebioda, Agata Kot-Wasik, Jakub Karczewski, and Magdalena Śliwka-Kaszyńska. 2018. "Chromatographic and Spectroscopic Identification and Recognition of Natural Dyes, Uncommon Dyestuff Components, and Mordants: Case Study of a 16th Century Carpet with Chintamani Motifs" Molecules 23, no. 2: 339. https://doi.org/10.3390/molecules23020339
APA StyleOtłowska, O., Ślebioda, M., Kot-Wasik, A., Karczewski, J., & Śliwka-Kaszyńska, M. (2018). Chromatographic and Spectroscopic Identification and Recognition of Natural Dyes, Uncommon Dyestuff Components, and Mordants: Case Study of a 16th Century Carpet with Chintamani Motifs. Molecules, 23(2), 339. https://doi.org/10.3390/molecules23020339