Interplay between P-Glycoprotein Expression and Resistance to Endoplasmic Reticulum Stressors
by
 and
and
Milan Hano
1, 
Lenka Tomášová
2,
Mário Šereš
1,
Lucia Pavlíková
1,
Albert Breier
3,* and
Zdena Sulová
1,* 1
Institute of Molecular Physiology and Genetics, Centre of Bioscience, Slovak Academy of Sciences, Dúbravska cesta 9, 84505 Bratislava, Slovakia
2
Institute of Clinical and Translational Research, Biomedical Research Center, Slovak Academy of Sciences, Dúbravska cesta 9, 84505 Bratislava, Slovakia
3
Institute of Biochemistry and Microbiology, Faculty of Chemical and Food Technology, Slovak University of Technology, Radlinského 9, 81237 Bratislava, Slovakia
*
Authors to whom correspondence should be addressed.
Molecules 2018, 23(2), 337; https://doi.org/10.3390/molecules23020337
Submission received: 13 January 2018
/
Revised: 30 January 2018
/
Accepted: 1 February 2018
/
Published: 6 February 2018
(This article belongs to the Special Issue Counteracting Drug Resistant Mechanisms in Cancer)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Multidrug resistance (MDR) is a phenotype of cancer cells with reduced sensitivity to a wide range of unrelated drugs. P-glycoprotein (P-gp)—a drug efflux pump (ABCB1 member of the ABC transporter gene family)—is frequently observed to be a molecular cause of MDR. The drug-efflux activity of P-gp is considered as the underlying mechanism of drug resistance against P-gp substrates and results in failure of cancer chemotherapy. Several pathological impulses such as shortages of oxygen and glucose supply, alterations of calcium storage mechanisms and/or processes of protein N-glycosylation in the endoplasmic reticulum (ER) leads to ER stress (ERS), characterized by elevation of unfolded protein cell content and activation of the unfolded protein response (UPR). UPR is responsible for modification of protein folding pathways, removal of misfolded proteins by ER associated protein degradation (ERAD) and inhibition of proteosynthesis. However, sustained ERS may result in UPR-mediated cell death. Neoplastic cells could escape from the death pathway induced by ERS by switching UPR into pro survival mechanisms instead of apoptosis. Here, we aimed to present state of the art information about consequences of P-gp expression on mechanisms associated with ERS development and regulation of the ERAD system, particularly focused on advances in ERS-associated therapy of drug resistant malignancies.
1. Introduction
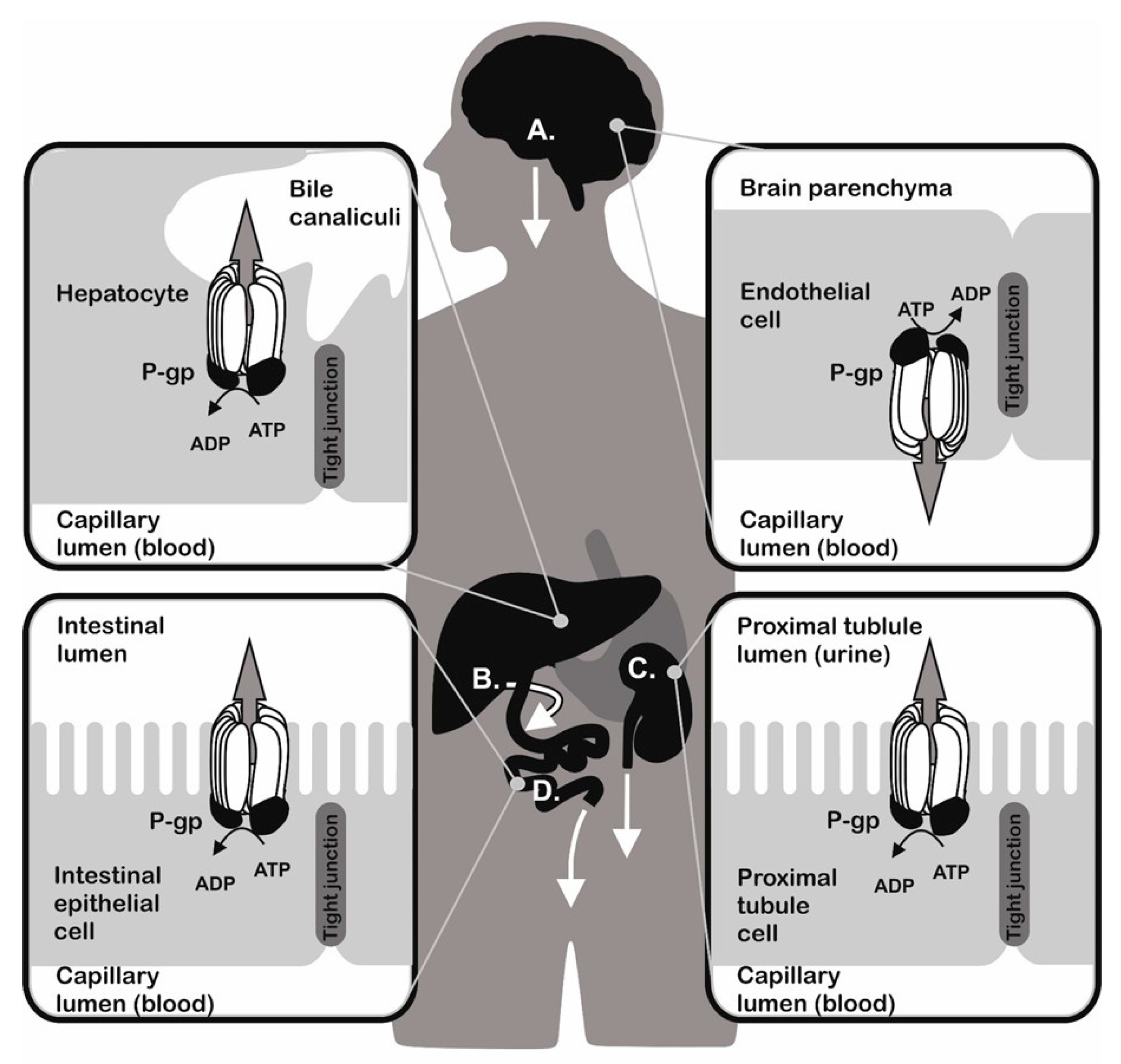
Organisms were persistently exposed to environmental attacks represented by different chemicals during evolution. To survive the unfavourable conditions, they developed functions to detoxify and remove toxins. Effective detoxification mechanisms are present both in unicellular (like bacteria and yeast) and multicellular (like plants and animals) organisms. These mechanisms are responsible for formation of defence systems against a broad spectrum of structurally unrelated substances that induce cell damage by different mechanisms [1,2,3,4,5,6,7,8,9,10]. The ATP Binding Cassette (ABC) transporters were proposed as universal detoxifiers, since they are present in a wide variety of organisms. They either transport substances out of cells or into the lumen of intracellular organelles. P-glycoprotein (P-gp, an ABCB1 member of the ABC transporter family) is encoded by the MDR1 (ABCB1) gene [11] and represents the first discovered [12] and most studied ABC transporter. The tissue distribution of P-gp is linked to its function as a transporter responsible for excretion of different compounds (Figure 1). P-gp is present at a higher amount in: (i) mucosal cells of the small intestine [13]; (ii) in the endothelial cells either of the blood brain barrier [14] or blood placental barrier [15], where it prevents the absorption of toxins; and (iii) in the kidney proximal tubule and hepatocytes, where it pumps metabolites and xenobiotics into the urine (substances that can be dissolved in urine) and bile (substances with limited solubility in water milieu) [16]. The transport function of P-gp protects cells against accumulation of harmful substances and thus plays an important role in maintaining physiological homeostasis. On the other hand, P-gp secured elimination of drugs, particularly anticancer agents, from the inner space of cells leads to the loss of pharmacological responses and development of multidrug resistance (MDR) to P-gp substrates. This phenomenon may result in chemotherapy failure in patients and consequent impairment of therapy outcome [17,18]. Similarly, analogues of P-gp either in bacteria [19] or in protozoa [20] were described to reduce sensitivity to antibiotics and anti-malarial drugs, respectively.
2. P-Glycoprotein and Multidrug Resistance (MDR)
Cancer cells can develop specific phenotypes characterized by overexpression of P-gp that confers resistance to a wide range of structurally unrelated substances belonging to a cluster of P-gp substrates. When expressed in neoplastic cells, P-gp can cause massive drug resistance to substrates involving anthracyclines (e.g., doxorubicin), vinca alkaloids (e.g., vincristine), actinomycines (e.g., actinomycin D, dactinomycines), taxols (e.g., paclitaxel), alkylating agents (mitomycin C), peptide antibiotics (gramicidin, valinomycin) and many others (reviewed in [23]). This phenotype could be inherently reflecting special functions of tissue from which neoplastic cells were developed or acquired due to cancer cell selection/adaptation to the presence of anticancer agents [17]. The resistant cells exhibit lower intracellular concentrations of anticancer drugs, which is related to alterations in the plasma membrane, particularly extremely improved drug efflux activity of P-gp that confers resistance to P-gp substrates by several hundred times [2,24]. Upregulation of the MDR1 gene is found in various cancer types [2,24,25,26,27,28,29,30,31,32]. The drug-efflux activity of P-gp is considered to be the underlying mechanism of MDR [12,33,34]. In addition, direct inhibition of tumour cell apoptosis was proposed in P-gp positive cells [35]. Alteration of apoptosis induced by drugs in P-gp positive cells have been described by several authors [36,37,38,39,40]. This activity is independent on P-gp drug efflux activity since transport-defective mutant P-gp expressed in CEM lymphoma cells suppresses vincristine-induced apoptosis via reduction of mitochondrial cytochrome C release and depressed caspase activation [41]. Moreover, we describe depression of cisplatin sensitivity (a substance that is not a P-gp substrate) in L1210 cells expressing P-gp due to either selection with vincristine or transfection with a human MDR1 gene [42,43]. P-gp via this antiapoptotic activity could induce significant cell resistance against substances that are not P-gp substrates.
P-gp is a polypeptide consisting of 1280 amino acid organized in two halves. Both halves have a strong structural similarity and contain a transmembrane domain formed by 6 α-helical membrane spans and an ATP binding site with ABC structural consensus (reviewed in [24]). After the binding of drugs to the intracellular P-gp drug binding domains oriented either to cytosol or inner membrane space, an ATP dependent conformation change of P-gp occurs and the agents are relocated to the extracellular space [44]. P-gp is synthetized on rough ER as a 150 kDa polypeptide precursor, which is after correct folding with calnexin and Hsc70 [45] further glycosylated into a 170 kDa mature protein [46,47]. P-gp moves from the ER to the Golgi apparatus (GA) for glycosylation and is afterwards incorporated into the plasma membrane. The regulation of P-gp trafficking from the ER to the plasma membrane is not completely clear. It was reported that microtubules are required for its transport from ER to GA [48] and a direct or indirect path to the plasma membrane via an intracellular endosome pool has been proposed [49,50]. Disruption of folding or glycosylation of glycoproteins (including P-gp) may lead to rapid proteasome-mediated degradation [51].
3. Protein Quality Control in Endoplasmic Reticulum (ER)
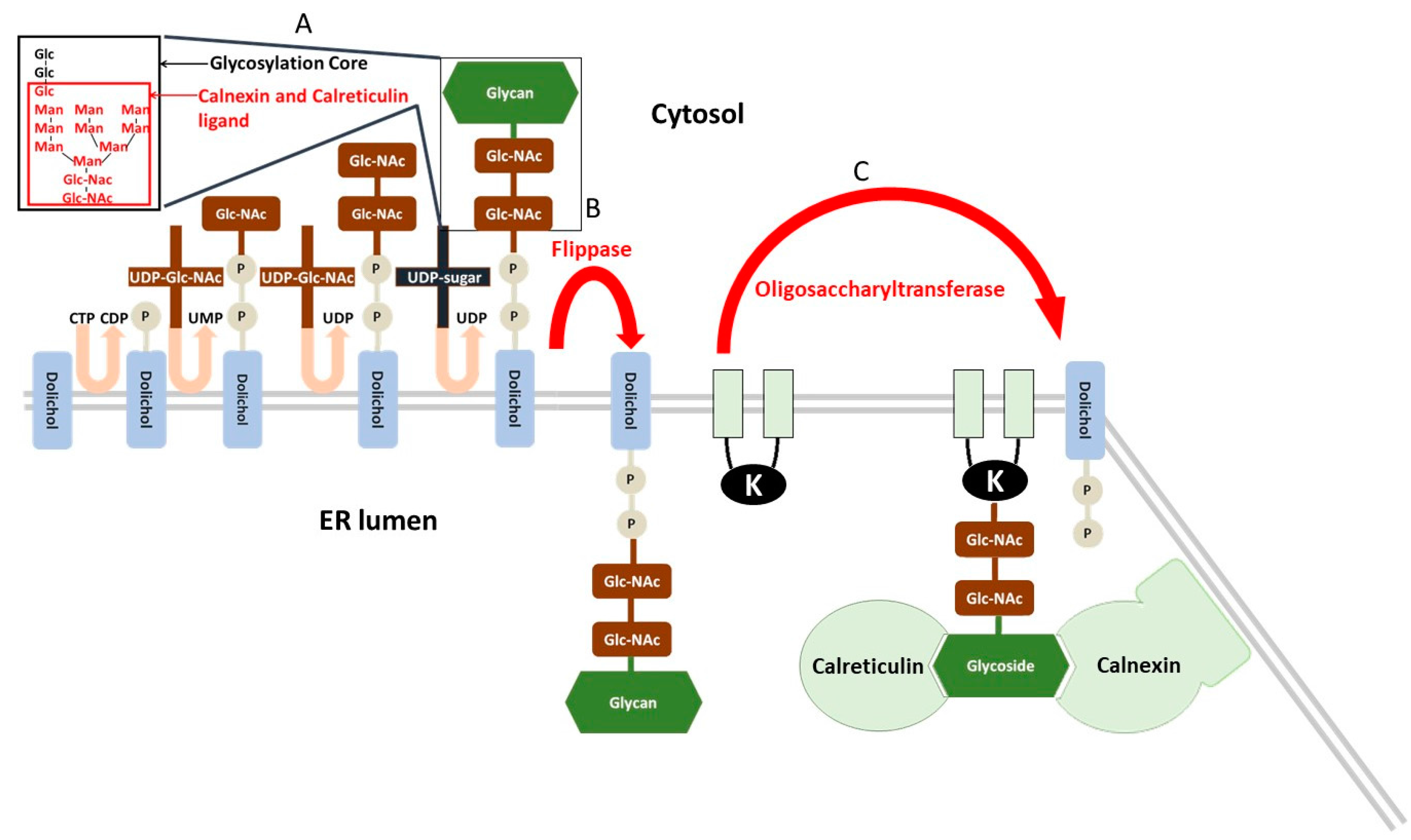
The endoplasmic reticulum (ER) is an organelle that secures cell homeostasis via serving the following functions: (i) proteosynthesis on ribosomes attached to rough ER; (ii) control of protein posttranslational modification, their folding and intracellular translocation; and (iii) storage of cell calcium and regulation of calcium homeostasis. In the case of correct folding, proteins enter the secretory pathway in the ER and GA [52]. N-glycosylation is the crucial step in the posttranslational modification in ER and represents a basic protein quality control [53]. The N-glycosylation is initiated in the ER while the protein is folded. Further processing of the N-glycan is catalysed by specific glycosidases and glycosyltransferases [54,55,56,57]. The elongation of the N-glycans and the O-glycosylation proceeds in the GA after the folding. First, the glycoside core (Glc3Man9NAcGlc2) linked with a dolichol phosphate anchored in the ER membrane is synthesized on the cytosolic side and flipped to the luminal side of the ER [58]. The glycoside core has a specific structure (documented in Figure 2) with three terminal glucoses [59]. After synthesis, the glycosylation core is relocated to the NH2 group of the asparagine residue of proteins undergoing N-glycosylation. Before the translocation to the GA, two chaperone proteins, the soluble calreticulin and the membrane bound calnexin, control the state of protein folding [60,61]. These lectins/chaperones exert Ca2+-dependent affinity to structure of glycosylation core with the one terminal glucose (GlcMan9NAcGlc2). Only properly folded proteins can escape from binding with calnexin and calreticulin and exit the ER.
Exiting the ER is secured by specific elimination of terminal glucoses with α-glucosidase I and II and consequent loss of ligand property for calnexin and calreticulin [59]. Unfolded proteins are reglucosylated by the UDP glucose:glycoprotein glucosyltransferase (UGGT) and re-enter the calnexin/calreticulin cycle several times to complete the folding [52]. However, persistently misfolded proteins are recognized by mannosidases, which remove mannose residues from the glycan [65,66] and thus prevent the reglucosylation by UGGT [67] and further calnexin/calreticulin protein quality control [52]. This is the first step of the ER-associated protein degradation (ERAD). After binding to the ERAD lectins, osteosarcoma amplified 9 (OS-9) or XTP3 transactivated protein (XTP3-B or erlectin), the deglycosylated proteins are removed to the cytoplasm and delivered for ubiquitination [68,69]. The transmembrane ubiquitin E3 ligase proteins link the recognition of misfolded proteins in the ER and the proteasome mediated degradation in the cytosol [70]. In the mammals, Hrd1/Suppressor of Lin-12-like (Sel1L) ligase is one of the ligases responsible for ubiquitination followed by degradation in proteasomes [71]. The Hrd1/Sel1L ligase is part of a complex with several other proteins (Derlin 1–3 proteins, p97 ATPase, VIMP, Herp), which extract and dislocate unfolded proteins from the ER membrane to the cytosol [72,73,74].
4. ER Stress, Induction and Consequences
Under ER stress (ERS) unfolded proteins accumulate in the lumen of the ER, resulting in activation of the unfolded protein response (UPR). The UPR program plays a crucial role in the regulation of the cell survival/death switch [75]. The primary action of the UPR is the modification of pathways responsible for protein folding, removal of misfolded proteins by ERAD and inhibition of protein synthesis in order to preserve the cell against stress induced by an excess of unfolded proteins [76]. However, in the case of sustained stress, UPR induces cell death [75]. Three ERS sensors control the UPR: inositol-requiring enzyme 1α (IRE1α) [77], pancreatic ER kinase (PERK) [78] and activating transcription factor 6 (ATF6) [79]. The activation of these three sensors under non-stress conditions is inhibited by the immunoglobulin binding protein (BiP), known also as glucose regulated protein 78 (GRP78). This protein is an intracellular chaperone with function either in correct folding of nascent polypeptides in ER or UPR regulation, which may protect cells against apoptosis induced by immature protein accumulation in ER. BiP is also known as a member of the heat-shock protein (HSP) 70 family and is upregulated in cells under oxygen and glucose limitation [80].
Accumulation of the unfolded proteins leads to the binding of the BiP to the unfolded proteins, due to a higher affinity of the BiP to unfolded proteins compared to ERS sensors. The dissociation of BiP from the luminal domains of stress sensors results in homodimerization of both IRE1α and PERK, their trans/auto phosphorylation, the translocation of ATF6 to the GA and subsequent activation [52,75].
The mechanisms of switching between cell survival and cell death under ERS and its regulation is not fully understood. The activated ERS sensors promote expression of both pro survival and pro apoptotic mediators [81]. It is believed that there is a mutual regulation between death-inducing and life-sustaining factors. Particularly in the early stage of ERS, the death inducing stimuli are antagonized by pro survival stimuli. On the contrary, long-lasting stress induces an opposite response, shifting the balance in favour of cell death [75] by the following mechanisms:
- The transcription factor X-box-binding protein (XBP1) regulates protein folding, trafficking and ERAD. After IRE1α induction of specific splicing of XBP1 mRNA, spliced variant sXBP1 enters the nucleus and regulates the expression of downstream products as UPR target genes, such as those encoding chaperones and components of ERAD [82,83,84]. The early pro survival response inhibits the expression of pro apoptotic C/EBP homologous protein (CHOP). However, prolonged ERS is associated with an increase of ROS levels and IRE-1α/XBP1 mediated activation of both CHOP and Bim and inhibition of Bcl-2, which leads to apoptosis [85,86,87,88,89].
- Activation of the PERK pathway leads to phosphorylation of eukaryotic initiation translation factor 2α (eIF2α), which triggers an antioxidant, pro survival response mediated by transcription factor 4 (ATF4). These phosphorylation leads to inhibition of translation [90]. In the case of sustained PERK stimulation, ATF4 signalling pathway induces dephosphorylation of eIF2α and promotes the expression of CHOP to mediate ERS-induced apoptosis [91,92].
- The ERS sensor ATF6 moves after activation from the ER to the GA, where it is cleaved by site 1 and site 2 proteases (S1P and S2P) to generate a cytosolic active transcription factor regulating protein folding and degradation [93].
The repression of the transcription factor E2F1, a direct inhibitor of apoptosis, was proposed as the crucial step in determining the death program [75]. It was reported that the late IRE1α/XBP1 response positively regulates the E2F1 gene and activates ATF6. The combined activity of E2F7 and ATF6 results in downregulation of E2F1 followed by a rapid apoptotic response [94].
ER stress may be pharmacologically induced by substances that alter the proper function of the ER [95]:
- Inhibitors of protein N-glycosylation, such as tunicamycin, an inhibitor of the UDP-N-acetylglucosamine-dolichol phosphate N-acetylglucosamine-1-phosphate transferase, that realize the first step of glycosylation core synthesis (Figure 2).
- Inducers of calcium depletion of ER, such as thapsigargin, that block the calcium pump of this organelle. Lack of calcium content in the ER eliminates proper function of calnexin and calreticulin and induces malfunction of protein quality control in the ER.
- Inhibitors of transport of proteins from the ER to the GA, such as brefeldin A, that additionally induce retrograde protein transport from the GA to the ER. This leads to the accumulation of unfolded proteins in the ER.
- Strong reducing agents, such as DTT, that block disulphide-bond formation and induce ERS within minutes.
- Proteasome inhibitors, such as, MG132 that block ERAD and cause misfolded protein accumulation in the ER.
5. ER Stress, Cancer and MDR
Uncontrolled proliferation of cancer cells leads to fast tumour growth associated with a low nutrient and oxygen supply that may induce disruption of cell homeostasis and ERS. Despite sustained activation of stress sensors, malignant cells do not switch to apoptosis. In contrast, they are able to adapt to ERS and deregulate the UPR in favour of cell survival, resulting in tumour development and progression [75,96]. Angiogenesis plays a crucial role in the progression of tumours by developing new vascular networks to supply nutrients and oxygen for malignant tissues [97]. All three sensors, PERK, IRE1α and ATF6, were reported to promote the expression of pro angiogenic vascular endothelial growth factor (VEGF) [98,99,100,101,102]. Moreover, neoplastic cells express increased levels of antioxidative factors as protection against the action of reactive oxygen species (ROS) [103]. In this context, PERK mediates the antioxidative nuclear factor-erythroid 2-related factor 2 (Nrf2) pathway and thus promotes the glutathione mediated buffer capacity of the generated ROS [104]. Similarly, activation of PERK was associated with tumour initiation and expansion by maintaining redox homeostasis and protecting the cancer cells from oxidative DNA damage [105]. Inhibitor of apoptosis-stimulating protein p53 (iASPP), a key inhibitor of this tumour suppressor, facilitates tumour growth by promotion of autophagy dependent on mTOR (mechanistic target of rapamycin) [106]. This p53 inhibitor is an antioxidative factor and drives cancer growth and drug resistance by competing with Nrf2 for keap1 (Kelch-like ECH-associated protein 1) binding [103].
The ERS sensors were suggested to play a role in tumour development. Low glucose levels and hypoxia in cancer cells resulted in activation of PERK and XBP1 and downstream pro survival pathways [107,108,109,110]. Several cancer types overexpress XBP1 and CHOP factors, related to higher cell proliferation and poor patients’ prognosis [111,112,113,114,115,116,117]. A transcriptional XBP1/HIF-1α complex was suggested to promote the tumorigenicity and progression of an aggressive subtype of human breast cancer [118]. Finally, overexpression of XBP1s was reported to play a role in the pathogenesis of multiple myeloma and a poor prognosis [119,120]. Conversely, several reports support the anti-tumour role of ERS, e.g., either better clinical prognosis observed in acute myeloid leukaemia patients with induced sXBP1 mRNA [121] or increased proliferation and malignant transformation after inhibition of the PERK/eIF2α pathway [122,123]. This could reflect the specificity of pro-/anti-tumour actions of ERS in different stages and types of malignant tissues.
Several members of the ERAD program have been proposed to play a role in the development of cancer [70]. Increased expression of Sel1L associated component of ubiquitination ligase was detected in different cancer types, which was related to reduced tumour growth and prolonged overall survival [124,125,126,127]. In contrast, glioblastoma multiform cells with a single nucleotide polymorphism rs12435998 genotype downregulated Sel1L, which was associated with a better prognosis of patients [128]. This polymorphism was proposed as a predictor of glioblastoma survival and response to radio-chemotherapy. It was suggested that ER lectin OS9 (known also as ERLEC2) promotes the tolerance of cancer cells to hypoxia by suppressing transcription of and mediating degradation of HIF 1α factor [129,130]. Furthermore, Sha et al. proposed that OS9 serves as a substrate for the Sel1L/Hrd1 complex [131]. It has to be stressed that regulation between OS9 and Sel1L in cancer cells needs further investigation. An ATP-dependent process of extracting the ubiquitinated proteins from the ER membrane is mediated by the p97 ATPase [132]. Inhibition of the p97 ATPase is related to increased apoptosis of cancer cells [133,134,135]. In addition, increased expression of p97 ATPase in B cell lymphomas has been reported; however, ERS was not activated [133]. In contrast, primary lung adenocarcinoma patients expressed lower levels of p97 ATPase, which induced ERS [136].
Recent evidence suggests that ERS is involved in the regulation of resistance to chemotherapy either independent or dependent on P-gp [137,138,139,140,141]. Cagnetta et al. reported that P-gp inhibitors induced ERS and ERS blockage strongly reduced the cytotoxic effect of the treatment in leukemic cells [137]. The mechanism underlying the induction of ERS by P-gp inhibition is not completely clear. However, the fact that insulin resistance promotes PERK mediated ERS, expression of Bcl-2 and P-gp in human hepatocarcinoma cells [138] indicated on strong relations between these ESR and P-gp function. In addition, expression of PERK in ERS resistant cells was increased and resulted in Nrf2 dependent transcription of the MRP1 gene. The ERS and chemotherapy resistance were reversed by disrupting the PERK/Nrf2 axis [141]. Interestingly, the expression of the other ERS sensors IRE1α and ATF6 were not altered in resistant cells [140]. In contrast, downregulation of the ATF6 pathway promoted cell death and reversed the resistance of the dormant tumour cells to rapamycin and glioblastoma cells to radiation [142,143]. The transport activity of P-gp is considered to be the underlying mechanism of imatinib resistance in chronic myeloid leukaemia [144]. Kusio Kobialka et al. showed that PERK/eIF2α phosphorylation was associated with chronic myeloid leukaemia progression and imatinib resistance. Furthermore, imatinib-mediated apoptosis downregulated PERK/eIF2α phosphorylation [145].
The C/EBP family of proteins is a group of transcription factors involved in regulation of cellular responses to ERS [146]. Riganti et al. investigated the role of C/EBP-β and CHOP members in establishing MDR. Transcription factor C/EBP-β can have a pro apoptotic effect, which is mediated by the natural dominant negative, truncated transcriptional repressor liver-enriched inhibitory protein LIP isoform. On the other hand, the liver-enriched transcriptional activator protein LAP isoform also promotes tumour progression by attenuating ERS-triggered cell death [147]. They determined that MDR cells do not express LIP, which undergoes ubiquitin-mediated degradation and failed to activate the pro apoptotic CHOP/caspase-3 pathway upon ERS or chemotherapy induction. They proposed that the lack of LIP results in two independent actions, particularly in upregulation of P-gp and attenuation of ERS triggered apoptosis [140].
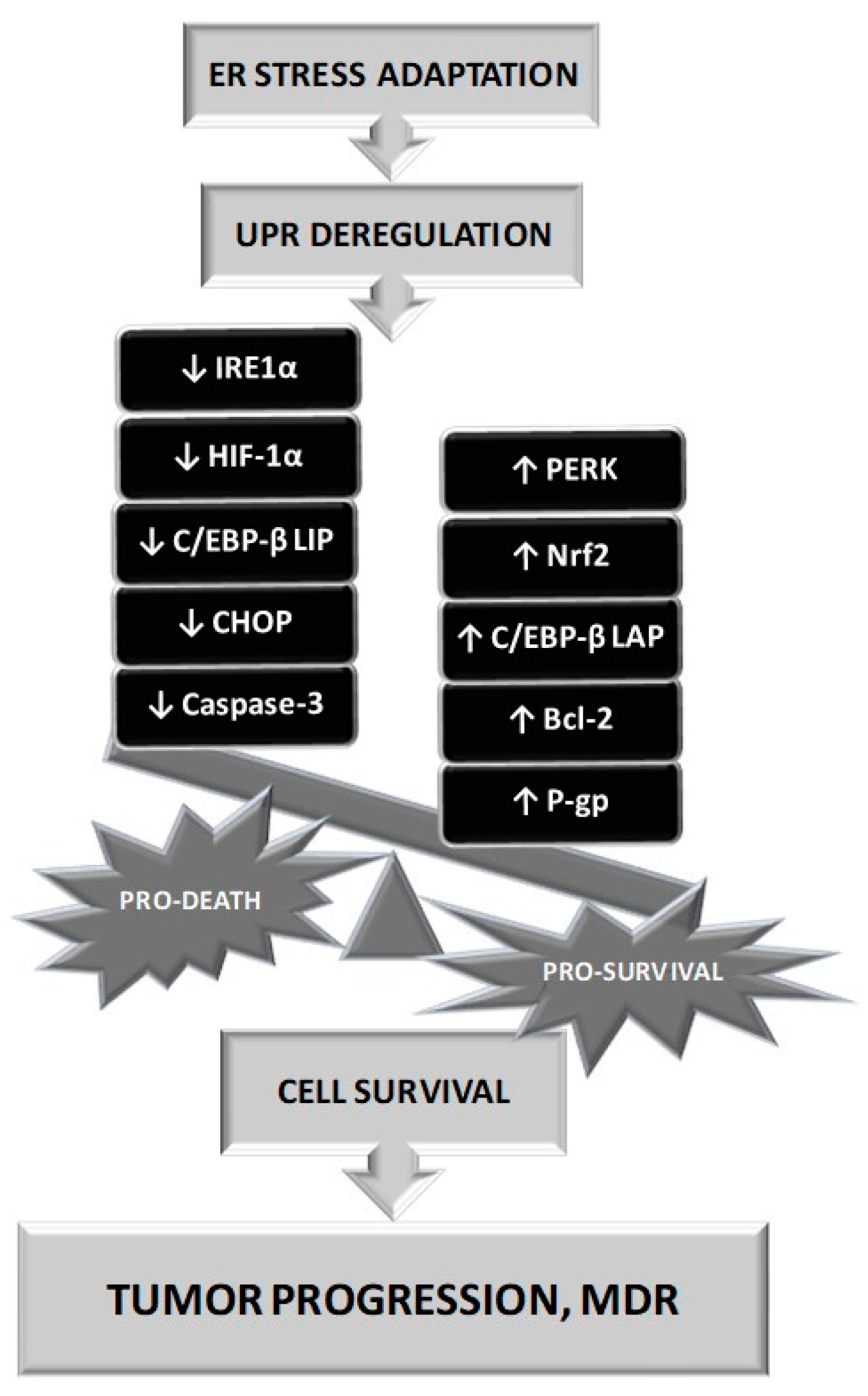
There also exists a gentle equilibrium between pro survival and pro death stimuli under ERS that is influenced by the severity of the stress and the duration of its presence. Under prolonged ERS, adaptation to this situation requires several steps and UPR deregulation may induce an imbalance of pro-survival mechanisms up to pro-death initiation and cells could escape from the death machinery induced by this situation (Figure 3).
6. Therapeutic Approaches
Firstly inhibition of P-gp with substances, which may be applied together with anticancer agents represents an important possibility to reduce P-gp antagonism against efficacy of cancer patients chemotherapy [148]. Recently, a new P-gp inhibitor tariquidar was developed as a product of rational drug design [149]. Tariquidar represents a high affinity, uncompetitive P-gp inhibitor that in contrast to verapamil, cyclosporine A and their analogues cannot be transported by P-gp [150]. This substance and its analogues are under intensive research to optimize the effective protocols for multidrug resistant malignancy treatment. For example, tariquidar in combination with doxorubicin, docetaxel, or vinorelbine in children and adolescents with recurrent or refractory solid tumours was tested clinically and a tolerable and biologically active dose of tariquidar was established [151].
Secondly targeting the UPR pathways in cancer cells may lead to a higher survival rate and reduced resistance of the cells to chemotherapy. Furthermore, the expression of ERS-mediated transcription factors may prove useful as prognosis indicators. The ERS inducers thapsigargin, tunicamycin and brefeldin A accelerated tumour growth in mice and human cancer cells [141,152]. In contrast, tunicamycin reduced tumour growth either in triple negative or double negative breast cancer models [153]. Cell treated by specific inhibitors of the UPR downstream mediators or mice malignant cells lacking IRE1α, XBP-1, PERK, or ATF6 are more sensitive to hypoxia, with higher production of ROS, lower angiogenesis, delayed tumour progression and metastasis [139,154,155,156,157,158,159,160,161,162,163]. Several inhibitors of IRE1α were identified in the last decade [159,164,165,166,167,168,169,170]. Inhibition of the IRE1α/XBP-1 pathway by toyocamycin, salicylaldehyde analogues and STF 083010 induced apoptosis in various tumours [167,168,169]. In addition, two new IRE1α inhibitors (4μ8C and KIRAs) that act allosterically were developed [159,164]. Two small molecules (GSK2656157 and GSK2606414) as selective PERK inhibitors were proposed [154,156]. Morrow et al. showed that pegylated-human-arginase I induced lymphoblastic T-cell leukaemia apoptosis by inhibiting the phosphorylation of eIF2α [171]. Furthermore, chemical chaperones, tauroursodeoxycholic acid and 4-phenyl butyric acid, which alleviate ERS, reduced tumour growth and tumorigenesis [152,170]. Proteasome inhibitors, bortezomib and toyocamycin, were also reported to suppress the PERK and IRE1α pathways, leading to increased apoptosis of cancer cells [160,172]. The cytotoxic effect of the proteasome inhibitors was promoted in combination with the p97 ATPase inhibitor eeyarestatin I [173,174]. In addition, IRE1α and ATF6 silencing promoted the apoptotic effect of rhabdovirus-mediated oncolysis [175]. On the contrary, valproic acid, a histone deacetylase inhibitor that is another promising chemotherapeutic agent, induced UPR by upregulating the expression of BiP, CHOP and Sel1L in glioma stem cells [176] and the chemical chaperone 4-phenyl butyric acid, an ERS inhibitor, reduced the cytotoxic effect of chemotherapeutics [137]. Furthermore, lower expression of Sel1L in glioma stem cells with the SNP rs12435998 genotype was related to enhanced sensitivity to valproic acid [128].
Several studies recently investigated the link between ERS and the resistance of cells to chemotherapy (Table S1 in Supplementary Files). Both activation and inhibition of ERS sensors were proposed to regulate the development of MDR. HIV protease inhibitors, which blocked ERS, decreased the transport activity of P-gp resulting in accumulation of berberine in macrophages [177]. Similarly, reduced tumour growth and restored chemosensitivity in resistant tumours were observed after PERK silencing [141,145]. On the other hand, the increased activation of PERK was associated with upregulation of P-glycoprotein and resistance to adriamycin in hepatocarcinoma cells [178]. Furthermore, the inhibition of IRE1α significantly improved the efficacy of oncolytic virus therapy in resistant tumour models [175].
Several studies recently investigated the link between ERS and the resistance of cells to chemotherapy (Table S1 in Supplementary Files). Both activation and inhibition of ERS sensors were proposed to regulate the development of MDR. HIV protease inhibitors, which blocked ERS, decreased the transport activity of P-gp resulting in accumulation of berberine in macrophages [177]. Similarly, reduced tumour growth and restored chemosensitivity in resistant tumours were observed after PERK silencing [141,145]. On the other hand, the increased activation of PERK was associated with upregulation of P-glycoprotein and resistance to adriamycin in hepatocarcinoma cells [178]. Furthermore, the inhibition of IRE1α significantly improved the efficacy of oncolytic virus therapy in resistant tumour models [175].
Chakravarty et al. [179] have investigated the effect of nelfinavir, an HIV protease inhibitor (also known as P-gp substrate [180]) on doxorubicin toxicity in an MDR breast cancer cell line. They reported that single exposure to nelfinavir transiently induced P-gp levels; however, multiple treatments with nelfinavir inhibited both P-gp expression and efflux activity together with activation of the pro apoptotic PERK/ATF4/CHOP pathway. Another study described the inhibition of XBP-1 in multiple myeloma cells, which induced bortezomib resistance [181]. Furthermore, rat C6 glioma cells developed resistance to emodin, an antitumor agent, which was related to overexpression of MDR genes and reduced ERS [182]. Thapsigargin, an ERS inducer, decreased the expression of P-gp in leukaemia cells and treatment with cyclosporine A, a P-gp inhibitor, increased expression of IRE1α [137,183]. Similarly, increased chemosensitivity of melanoma stem-like cells was associated with induction of ERS [184]. Finally, Song et al. reported that ERS reduced drug resistance in breast cancer; however, the transport activity of P-gp was not altered [185].
Tunicamycin, blocks protein N-glycosylation and thus leads to the accumulation of unfolded proteins and the activation of ERS [186,187]. It was reported that inhibition of N-glycosylation, a major posttranslational modification of P-gp, by tunicamycin caused rapid ubiquitination and proteasome-dependent degradation of P-gp [188,189]. Furthermore, Kramer et al. reported that tunicamycin reversed P-gp mediated MDR [190]. In contrast, our studies showed that tunicamycin failed to reverse the efflux activity of P-gp in leukaemia cells, suggesting that unglycosylated P-gp has the ability to escape from the ERAD system and become functionally integrated into the plasma membrane [191,192,193]. This is in line with the Riganti et al. study, where tunicamycin did not alter the efflux activity or levels of P-gp, MRP1 or MRP2 [140]. Interestingly, unglycosylated P-gp that was found in the membrane and exerted P-gp efflux activity as measured by a calcein retention assay (after treatment with tunicamycin [193]) was ubiquitinated [191]. Thus, after ubiquitination, P-gp continued to mature instead of being degraded in proteasomes. Ubiquitin is a small polypeptide (76 amino acids, Mr 8500 kDa) that can conjugate with several proteins via formation of isopeptide bond between ubiquitin C-terminal glycine and lysine in target protein. Ubiquitin contains seven lysines (K6, K11, K27, K29, K33, K48 and K63) and any of them could form isopeptide bond with C-terminal glycine of another ubiquitin molecule to form polyubiquitin chain [194]. Therefore, ubiquitination produces either monoubiquitinated or polyubiquitinated proteins. Protein ubiquitination leads to increase of protein molecular weight reflecting the number of ubiquitins linked in polyubiquitin chains attached to protein [195]. Monoubiquitination seems to be followed by chromatin regulation, protein sorting and trafficking, whereas polyubiquitination is rather associated with protein degradation in proteasome [196]. In previous paper, we observed larger P-gp ubiquitination after treatment of P-gp positive variants of mouse leukaemia cell line L1210 with tunicamycin. This ubiquitination was associated with only small elevation in P-gp molecular weight, which excludes massive polyubiquitination [191]. Ubiquitinated and active P-gp was still localized in plasma membrane [193]. Beside number of ubiquitins in polyubiquitin chains also structural feature, i.e. which from seven lysines was used for isopeptide bond formation is important for final direction of ubiquitin signalling. For example, proteasomal degradation is typical consequence of protein polyubiquitination through lysine in position 48 on ubiquitin molecule (conventional chain). However, similarly abundant polyubiquitination utilizing K63 leads rather to kinase activation, DNA- repair and vesicle trafficking [197]. Taken together, the destiny of ubiquitinated proteins, after blockage of N-glycosylation and overall quality control in ER by tunicamycin, depends on the number of bound ubiquitins in the chain and, in the case of polyubiquitination, on special structural features of the polyubiquitin chain [191].
7. Conclusions
Cancer cells are able to take advantage of cell programs, which serve to maintain physiological function of the cells under stress conditions. Persistent ERS, due to different pathological impulses including hypoxia and glucose shortage in the tumour environment, or the presence of ER stressors, should result in cell death. However, cancer cells are able to stay in an extremely prolonged early pro survival response to ERS. Moreover, cancer cells can induce expression of P-gp and use its efflux activity for removal of xenobiotics to survive the cytotoxic effect of chemotherapy. The mechanism of how malignant cells adapt to ERS and overcome the UPR cell death program is crucial for understanding the progression and resistance of tumours to therapy. One of the possibilities is that adaptation to ERS by cancer cells may lead to overexpression and induction of P-gp and thus contribute to production of an MDR phenotype. It seems that increases of PERK activity, an ERS sensor and loss of C/EBP LIP in resistant cells results in overexpression of P-gp. Furthermore, modification of the ERAD system can be observed in the case of unglycosylated P-gp (due to N-glycosylation blockage by tunicamycin [193]), which after ubiquitination could escape from the proteasomal degradation cascade and in ubiquitinated form is integrated into the plasma membrane and maintains its function [191]. Therefore, targeting the UPR pathways in cancer cells may lead to disturbed P-gp function and reverse resistance of the cells to chemotherapy. These facts suggest a new role of ERS sensors and ERAD components as therapeutic targets in the treatment of resistant tumours. In addition, mutation of the ERS sensor genes, particularly the missense mutations enriched in PERK, have been proposed to be responsible for the changes in UPR [198,199]. However, mutations differ among various types of cancers [200]; thus, characterization of tumour specific mutations and the impact of ERS adaptation in the relationship to P-gp dependent MDR is needed to verify the above hypotheses.
Supplementary Materials
The following are available online.
Acknowledgments
Our laboratories were financially supported by grants from the Slovak APVV grant agency (No. APVV-14-0334, APVV-15-0303), the VEGA grant agency (2/0028/15, 2/0156/16, 2/0122/17) and a Building Infrastructure for Modern Research of Civilization’s Diseases project (ITMS 26230120006). The paper was edited for proper English language, grammar, punctuation, spelling and overall style by one or more of the highly qualified native English-speaking editors at American Journal Experts.
Author Contributions
Milan Hano prepared the manuscript draft; Milan Hano, Lenka Tomášová, Mário Šereš and Lucia Pavlíková prepared the literary research; Albert Breier and Zdena Sulová prepared the final version of the manuscript.
Conflicts of Interest
We declare that there are no conflicts of interest.
Abbreviations
| ATF6 | activating transcription factor 6 |
| ABC | ATP-binding cassette |
| BiP | immunoglobulin binding protein |
| CHOP | C/EBP homologous protein |
| eIF2α | eukaryotic initiation translation factor 2α |
| ER | endoplasmic reticulum |
| ERAD | ER associated protein degradation |
| ERS | ER stress |
| GA | golgi apparatus |
| GRP78 | glucose regulated protein 78 |
| HSP | heat-shock protein |
| iASPP | inhibitor of apoptosis-stimulating protein p53 |
| IRE1α | inositol-requiring enzyme 1α |
| keap1 | Kelch-like ECH-associated protein 1 |
| MDR | multidrug resistance |
| mTOR | mechanistic target of rapamycin |
| Nrf2 | nuclear factor-erythroid 2-related factor 2 |
| OS9 | osteosarcoma amplified 9 |
| PERK | pancreatic ER kinase |
| P-gp | P-glycoprotein |
| ROS | reactive oxygen species |
| S1P and S2P | site 1 and site 2 proteases |
| UGGT | UDP-glucose:glycoprotein glycosyltransferase |
| URP | unfolded protein response |
| VEGF | vascular endothelial growth factor |
| XBP1 | X-box-binding protein |
| XTP3 | lectin of ER |
| XTP3-B | transactivated protein or ER lectin |
References
- Borst, P.; Schinkel, A.H. Genetic dissection of the function of mammalian P-glycoproteins. Trends Genet. 1997, 13, 217–222. [Google Scholar] [CrossRef]
- Efferth, T.; Volm, M. Multiple resistance to carcinogens and xenobiotics: P-glycoproteins as universal detoxifiers. Arch. Toxicol 2017, 91, 2515–2538. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.; Costa, J.; Reis-Henriques, M.A. ABC transporters in fish species: A review. Front. Physiol. 2014, 5, 266. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, R.M. ABC multidrug transporters in schistosomes and other parasitic flatworms. Parasitol. Int. 2013, 62, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.M.; George, A.M. Multidrug resistance in parasites: ABC transporters, P-glycoproteins and molecular modelling. Int J. Parasitol. 2005, 35, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Kerr, I.D.; Jones, P.M.; George, A.M. Multidrug efflux pumps: The structures of prokaryotic ATP-binding cassette transporter efflux pumps and implications for our understanding of eukaryotic P-glycoproteins and homologues. FEBS J. 2010, 277, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Merola, V.M.; Eubig, P.A. Toxicology of avermectins and milbemycins (macrocylic lactones) and the role of p-glycoprotein in dogs and cats. Vet. Clin. North Am. Small Anim. Pract. 2012, 42, 313–333. [Google Scholar] [CrossRef] [PubMed]
- Myllynen, P.; Kummu, M.; Sieppi, E. ABCB1 and ABCG2 expression in the placenta and fetus: An interspecies comparison. Expert Opin. Drug. Metab. Toxicol. 2010, 6, 1385–1398. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.J. Too much of a good thing: How insects cope with excess ions or toxins in the diet. J. Exp. Biol. 2009, 212, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Goffeau, A. Yeast ATP-binding cassette transporters conferring multidrug resistance. Annu. Rev. Microbiol. 2012, 66, 39–63. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chin, J.E.; Ueda, K.; Clark, D.P.; Pastan, I.; Gottesman, M.M.; Roninson, I.B. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell 1986, 47, 381–389. [Google Scholar] [CrossRef]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in chinese hamster ovary cell mutants. Biochim. Biophys. Acta 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Thiebaut, F.; Tsuruo, T.; Hamada, H.; Gottesman, M.M.; Pastan, I.; Willingham, M.C. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 7735–7738. [Google Scholar] [CrossRef] [PubMed]
- Jolliet-Riant, P.; Tillement, J.P. Drug transfer across the blood-brain barrier and improvement of brain delivery. Fundam. Clin. Pharmacol. 1999, 13, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Molsa, M.; Heikkinen, T.; Hakkola, J.; Hakala, K.; Wallerman, O.; Wadelius, M.; Wadelius, C.; Laine, K. Functional role of P-glycoprotein in the human blood-placental barrier. Clin. Pharmacol. Ther. 2005, 78, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Mayer, U.; Wagenaar, E.; Mol, C.A.; van Deemter, L.; Smit, J.J.; van der Valk, M.A.; Voordouw, A.C.; Spits, H.; van Tellingen, O.; et al. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc. Natl. Acad. Sci. USA 1997, 94, 4028–4033. [Google Scholar] [CrossRef] [PubMed]
- Kvackajova-Kisucka, J.; Barancik, M.; Breier, A. Drug transporters and their role in multidrug resistance of neoplastic cells. Gen. Physiol. Biophys. 2001, 20, 215–237. [Google Scholar] [PubMed]
- Thomas, H.; Coley, H.M. Overcoming multidrug resistance in cancer: An update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control. 2003, 10, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ma, S. Efflux pump inhibitors: A strategy to combat P-glycoprotein and the nora multidrug resistance pump. ChemMedChem 2010, 5, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Ibraheem, Z.O.; Abd Majid, R.; Noor, S.M.; Sedik, H.M.; Basir, R. Role of different PFCRT and PFMDR-1 mutations in conferring resistance to antimalaria drugs in plasmodium falciparum. Malar. Res. Treat. 2014, 2014, 950424. [Google Scholar] [PubMed]
- Tomasova, L.; Konopelski, P.; Ufnal, M. Gut bacteria and hydrogen sulfide: The new old players in circulatory system homeostasis. Molecules 2016, 21, 1558. [Google Scholar] [CrossRef] [PubMed]
- Ufnal, M.; Pham, K. The gut-blood barrier permeability—A new marker in cardiovascular and metabolic diseases? Med. Hypotheses 2017, 98, 35–37. [Google Scholar] [CrossRef] [PubMed]
- Breier, A.; Barancik, M.; Sulova, Z.; Uhrik, B. P-glycoprotein—Implications of metabolism of neoplastic cells and cancer therapy. Curr. Cancer Drug. Targets 2005, 5, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Breier, A.; Gibalova, L.; Seres, M.; Barancik, M.; Sulova, Z. New insight into P-glycoprotein as a drug target. Anticancer. Agents Med. Chem. 2013, 13, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Kahng, J.; Kim, M.; Lim, J.; Kim, Y.; Cho, B.; Kim, H.K.; Min, W.S.; Kim, C.C.; Lee, K.Y.; et al. Expression of functional markers in acute nonlymphoblastic leukemia. Acta Haematol. 2000, 104, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Imrichova, D.; Messingerova, L.; Seres, M.; Kavcova, H.; Pavlikova, L.; Coculova, M.; Breier, A.; Sulova, Z. Selection of resistant acute myeloid leukemia SKM-1 and MOLM-13 cells by vincristine-, mitoxantrone- and lenalidomide-induced upregulation of P-glycoprotein activity and downregulation of CD33 cell surface exposure. Eur. J. Pharm. Sci. 2015, 77, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Messingerova, L.; Imrichova, D.; Kavcova, H.; Seres, M.; Sulova, Z.; Breier, A. A decrease in cellular microRNA-27a content is involved in azacytidine-induced P-glycoprotein expression in SKM-1 cells. Toxicol. In Vitro 2016, 36, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Messingerova, L.; Imrichova, D.; Kavcova, H.; Turakova, K.; Breier, A.; Sulova, Z. Acute myeloid leukemia cells MOLM-13 and SKM-1 established for resistance by azacytidine are crossresistant to P-glycoprotein substrates. Toxicol. In Vitro 2015, 29, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Polekova, L.; Barancik, M.; Mrazova, T.; Pirker, R.; Wallner, J.; Sulova, Z.; Breier, A. Adaptation of mouse leukemia cells L1210 to vincristine. Evidence for expression of P-glycoprotein. Neoplasma 1992, 39, 73–77. [Google Scholar] [PubMed]
- Roninson, I.B.; Chin, J.E.; Choi, K.G.; Gros, P.; Housman, D.E.; Fojo, A.; Shen, D.W.; Gottesman, M.M.; Pastan, I. Isolation of human mdr DNA sequences amplified in multidrug-resistant KB carcinoma cells. Proc. Natl. Acad. Sci. USA 1986, 83, 4538–4542. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.W.; Fojo, A.; Chin, J.E.; Roninson, I.B.; Richert, N.; Pastan, I.; Gottesman, M.M. Human multidrug-resistant cell lines: Increased MDR1 expression can precede gene amplification. Science 1986, 232, 643–645. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.B.; Lind, M.J.; Cawkwell, L. Establishment of in-vitro models of chemotherapy resistance. Anticancer. Drugs 2007, 18, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.R.; Ling, V. Purification of p-glycoprotein from plasma membrane vesicles of chinese hamster ovary cell mutants with reduced colchicine permeability. J. Biol. Chem. 1979, 254, 12701–12705. [Google Scholar] [PubMed]
- Gottesman, M.M.; Ling, V. The molecular basis of multidrug resistance in cancer: The early years of P-glycoprotein research. FEBS Lett. 2006, 580, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Zu, Y.; Yang, Z.; Tang, S.; Han, Y.; Ma, J. Effects of P-glycoprotein and its inhibitors on apoptosis in K562 cells. Molecules 2014, 19, 13061–13075. [Google Scholar] [CrossRef] [PubMed]
- Bear, C.E. Drugs transported by P-glycoprotein inhibit a 40 PS outwardly rectifying chloride channel. Biochem. Biophys. Res. Commun. 1994, 200, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.M.; Wei, L.Y.; Roepe, P.D. Are altered pHi and membrane potential in hu MDR 1 transfectants sufficient to cause mdr protein-mediated multidrug resistance? J. Gen. Physiol. 1996, 108, 295–313. [Google Scholar] [CrossRef] [PubMed]
- Idriss, H.T.; Hannun, Y.A.; Boulpaep, E.; Basavappa, S. Regulation of volume-activated chloride channels by p-glycoprotein: Phosphorylation has the final say! J. Physiol. 2000, 524, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Pallis, M.; Russell, N. P-glycoprotein plays a drug-efflux-independent role in augmenting cell survival in acute myeloblastic leukemia and is associated with modulation of a sphingomyelin-ceramide apoptotic pathway. Blood 2000, 95, 2897–2904. [Google Scholar] [PubMed]
- Ruefli, A.A.; Johnstone, R.W. A role for P-glycoprotein in regulating cell growth and survival. Clin. Appl. Immunol. Rev. 2003, 4, 31–47. [Google Scholar] [CrossRef]
- Tainton, K.M.; Smyth, M.J.; Jackson, J.T.; Tanner, J.E.; Cerruti, L.; Jane, S.M.; Darcy, P.K.; Johnstone, R.W. Mutational analysis of P-glycoprotein: Suppression of caspase activation in the absence of atp-dependent drug efflux. Cell Death Differ. 2004, 11, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Gibalova, L.; Sedlak, J.; Labudova, M.; Barancik, M.; Rehakova, A.; Breier, A.; Sulova, Z. Multidrug resistant P-glycoprotein positive L1210/VCR cells are also cross-resistant to cisplatin via a mechanism distinct from P-glycoprotein-mediated drug efflux activity. Gen. Physiol. Biophys. 2009, 28, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Gibalova, L.; Seres, M.; Rusnak, A.; Ditte, P.; Labudova, M.; Uhrik, B.; Pastorek, J.; Sedlak, J.; Breier, A.; Sulova, Z. P-glycoprotein depresses cisplatin sensitivity in l1210 cells by inhibiting cisplatin-induced caspase-3 activation. Toxicol. In Vitro 2012, 26, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F.; Gottesman, M.M. Is the multidrug transporter a flippase? Trends Biochem. Sci. 1992, 17, 18–21. [Google Scholar] [CrossRef]
- Gething, M.J.; Sambrook, J. Protein folding in the cell. Nature 1992, 355, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Greer, D.A.; Ivey, S. Distinct N-glycan glycosylation of P-glycoprotein isolated from the human uterine sarcoma cell line MES-SA/DX5. Biochim. Biophys. Acta 2007, 1770, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Thapsigargin or curcumin does not promote maturation of processing mutants of the abc transporters, CFTR, and P-glycoprotein. Biochem. Biophys. Res. Commun. 2004, 325, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Presley, J.F.; Cole, N.B.; Schroer, T.A.; Hirschberg, K.; Zaal, K.J.; Lippincott-Schwartz, J. ER-to-Golgi transport visualized in living cells. Nature 1997, 389, 81–85. [Google Scholar] [PubMed]
- Fu, D.; Roufogalis, B.D. Actin disruption inhibits endosomal traffic of P-glycoprotein-EGFP and resistance to daunorubicin accumulation. Am. J. Physiol.-Cell Physiol. 2007, 292, C1543–C1552. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, Y.; Dutt, P.; Lippincott-Schwartz, J.; Arias, I.M. Rab11a and myosin Vb are required for bile canalicular formation in WIF-B9 cells. Proc. Natl. Acad. Sci. USA 2005, 102, 15087–15092. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Groenendyk, J.; Michalak, M. Glycoprotein quality control and endoplasmic reticulum stress. Molecules 2015, 20, 13689–13704. [Google Scholar] [CrossRef] [PubMed]
- Sitia, R.; Braakman, I. Quality control in the endoplasmic reticulum protein factory. Nature 2003, 426, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Deprez, P.; Gautschi, M.; Helenius, A. More than one glycan is needed for er glucosidase ii to allow entry of glycoproteins into the calnexin/calreticulin cycle. Mol. Cell 2005, 19, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, J.Q.; Sharma, S.; Leister, K.J.; Joshi, L. A tight-knit group: Protein glycosylation, endoplasmic reticulum stress and the unfolded protein response. In Endoplasmic Reticulum Stress in Health and Disease; Agostinis, P., Afshin, S., Eds.; Springer: Dordrecht, The Netherlands, 2014. [Google Scholar]
- Pelletier, M.F.; Marcil, A.; Sevigny, G.; Jakob, C.A.; Tessier, D.C.; Chevet, E.; Menard, R.; Bergeron, J.J.; Thomas, D.Y. The heterodimeric structure of glucosidase ii is required for its activity, solubility, and localization in vivo. Glycobiology 2000, 10, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Shailubhai, K.; Pukazhenthi, B.S.; Saxena, E.S.; Varma, G.M.; Vijay, I.K. Glucosidase I, a transmembrane endoplasmic reticular glycoprotein with a luminal catalytic domain. J. Biol. Chem. 1991, 266, 16587–16593. [Google Scholar] [PubMed]
- Sanyal, S.; Frank, C.G.; Menon, A.K. Distinct flippases translocate glycerophospholipids and oligosaccharide diphosphate dolichols across the endoplasmic reticulum. Biochemistry 2008, 47, 7937–7946. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Cummings, R.D.; Esko, J.D.; Freeze, H.H.; Stanley, P.; Bertozzi, C.R.; Hart, G.W.; Etzler, M.E. Essentials of Glycobiology, 2nd ed.; CSH Laboratory Press: New York, NY, USA, 2009. [Google Scholar]
- Spiro, R.G.; Zhu, Q.; Bhoyroo, V.; Soling, H.D. Definition of the lectin-like properties of the molecular chaperone, calreticulin, and demonstration of its copurification with endomannosidase from rat liver golgi. J. Biol. Chem. 1996, 271, 11588–11594. [Google Scholar] [CrossRef] [PubMed]
- Ware, F.E.; Vassilakos, A.; Peterson, P.A.; Jackson, M.R.; Lehrman, M.A.; Williams, D.B. The molecular chaperone calnexin binds GLc1Man9GLcNAc2 oligosaccharide as an initial step in recognizing unfolded glycoproteins. J. Biol. Chem. 1995, 270, 4697–4704. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Dudek, J.; Gogala, M.; Schorr, S.; Linxweiler, J.; Lang, S.; Becker, T.; Beckmann, R.; Zimmermann, R.; Forster, F. Structure of the mammalian oligosaccharyl-transferase complex in the native er protein translocon. Nat. Commun. 2014, 5, 3072. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Groenendyk, J.; Szabo, E.; Gold, L.I.; Opas, M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 2009, 417, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.B. Beyond lectins: The calnexin/calreticulin chaperone system of the endoplasmic reticulum. J. Cell Sci. 2006, 119, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Tremblay, L.O.; You, Z.; Herscovics, A.; Wada, I.; Nagata, K. Enhancement of endoplasmic reticulum (ER) degradation of misfolded Null Hong Kong α1-antitrypsin by human er mannosidase I. J. Biol. Chem. 2003, 278, 26287–26294. [Google Scholar] [CrossRef] [PubMed]
- Ninagawa, S.; Okada, T.; Sumitomo, Y.; Kamiya, Y.; Kato, K.; Horimoto, S.; Ishikawa, T.; Takeda, S.; Sakuma, T.; Yamamoto, T.; et al. EDEM2 initiates mammalian glycoprotein ERAD by catalyzing the first mannose trimming step. J. Cell Biol. 2014, 206, 347–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, M.C.; Ferrero-Garcia, M.A.; Parodi, A.J. Recognition of the oligosaccharide and protein moieties of glycoproteins by the UDP-Glc:Glycoprotein glucosyltransferase. Biochemistry 1992, 31, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Christianson, J.C.; Shaler, T.A.; Tyler, R.E.; Kopito, R.R. OS-9 and GRP94 deliver mutant α1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008, 10, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Kikkert, M.; Doolman, R.; Dai, M.; Avner, R.; Hassink, G.; van Voorden, S.; Thanedar, S.; Roitelman, J.; Chau, V.; Wiertz, E. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 3525–3534. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Bhattacharya, A.; Qi, L. Endoplasmic reticulum quality control in cancer: Friend or foe. Semin. Cancer Biol. 2015, 33, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Tsai, B.; Ye, Y.; Rapoport, T.A. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat. Rev. Mol. Cell Biol. 2002, 3, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Lilley, B.N.; Ploegh, H.L. Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 14296–14301. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Standera, S.; Buerger, E.; Kikkert, M.; van Voorden, S.; Wiertz, E.; Koning, F.; Kloetzel, P.M.; Seeger, M. The ubiquitin-domain protein HERP forms a complex with components of the endoplasmic reticulum associated degradation pathway. J. Mol. Biol. 2005, 354, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Shibata, Y.; Kikkert, M.; van Voorden, S.; Wiertz, E.; Rapoport, T.A. Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 14132–14138. [Google Scholar] [CrossRef] [PubMed]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Z.; Harding, H.P.; Zhang, Y.; Jolicoeur, E.M.; Kuroda, M.; Ron, D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998, 17, 5708–5717. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Yao, J.; Zeng, W.; Mizuno, Y.; Kamm, K.E.; Stull, J.T.; Harding, H.P.; Ron, D.; Muallem, S. ER stress disrupts Ca2+-signaling complexes and Ca2+ regulation in secretory and muscle cells from PERK-knockout mice. J. Cell Sci. 2006, 119, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [PubMed]
- Panayi, G.S.; Corrigall, V.M. Immunoglobulin heavy-chain-binding protein (BiP): A stress protein that has the potential to be a novel therapy for rheumatoid arthritis. Biochem. Soc. Trans. 2014, 42, 1752–1755. [Google Scholar] [CrossRef] [PubMed]
- Vandewynckel, Y.P.; Laukens, D.; Geerts, A.; Bogaerts, E.; Paridaens, A.; Verhelst, X.; Janssens, S.; Heindryckx, F.; Van Vlierberghe, H. The paradox of the unfolded protein response in cancer. Anticancer. Res. 2013, 33, 4683–4694. [Google Scholar] [PubMed]
- Back, S.H.; Schroder, M.; Lee, K.; Zhang, K.; Kaufman, R.J. ER stress signaling by regulated splicing: IRE1/HAC1/XBP1. Methods 2005, 35, 395–416. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Yoshida, H.; Kokame, K.; Kaufman, R.J.; Mori, K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J. Biochem. 2004, 136, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Ahmed, H.; Yang, P.; Czinn, S.J.; Blanchard, T.G. Endoplasmic reticulum stress and IRE-1 signaling cause apoptosis in colon cancer cells in response to andrographolide treatment. Oncotarget 2016, 7, 41432–41444. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Banerjee, V.; Czinn, S.; Blanchard, T. Increased reactive oxygen species levels cause er stress and cytotoxicity in andrographolide treated colon cancer cells. Oncotarget 2017, 8, 26142–26153. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Hubbard, S.R. How IRE1 reacts to er stress. Cell 2008, 132, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Zeriouh, W.; Nani, A.; Belarbi, M.; Dumont, A.; de Rosny, C.; Aboura, I.; Ghanemi, F.Z.; Murtaza, B.; Patoli, D.; Thomas, C.; et al. Phenolic extract from oleaster (Olea europaea var. Sylvestris) leaves reduces colon cancer growth and induces caspase-dependent apoptosis in colon cancer cells via the mitochondrial apoptotic pathway. PLoS ONE 2017, 12, e0170823. [Google Scholar]
- Knutsen, J.H.; Rodland, G.E.; Boe, C.A.; Haland, T.W.; Sunnerhagen, P.; Grallert, B.; Boye, E. Stress-induced inhibition of translation independently of eIF2α phosphorylation. J. Cell Sci. 2015, 128, 4420–4427. [Google Scholar] [CrossRef] [PubMed]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eiF2α. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The role of the PERK/eiF2α/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process srebps. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Pagliarini, V.; Giglio, P.; Bernardoni, P.; De Zio, D.; Fimia, G.M.; Piacentini, M.; Corazzari, M. Downregulation of E2F1 during ER stress is required to induce apoptosis. J. Cell Sci. 2015, 128, 1166–1179. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Urano, F. Measuring er stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011, 490, 71–92. [Google Scholar] [PubMed]
- Shen, K.; Johnson, D.W.; Vesey, D.A.; McGuckin, M.A.; Gobe, G.C. Role of the unfolded protein response in determining the fate of tumor cells and the promise of multi-targeted therapies. Cell Stress Chaperones 2017, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Blais, J.D.; Addison, C.L.; Edge, R.; Falls, T.; Zhao, H.; Wary, K.; Koumenis, C.; Harding, H.P.; Ron, D.; Holcik, M.; et al. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol. Cell Biol. 2006, 26, 9517–9532. [Google Scholar] [CrossRef] [PubMed]
- Drogat, B.; Auguste, P.; Nguyen, D.T.; Bouchecareilh, M.; Pineau, R.; Nalbantoglu, J.; Kaufman, R.J.; Chevet, E.; Bikfalvi, A.; Moenner, M. IRE1 signaling is essential for ischemia-induced vascular endothelial growth factor-A expression and contributes to angiogenesis and tumor growth in vivo. Cancer Res. 2007, 67, 6700–6707. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Lipson, K.L.; Sargent, K.E.; Mercurio, A.M.; Hunt, J.S.; Ron, D.; Urano, F. Transcriptional regulation of VEGF-a by the unfolded protein response pathway. PLoS ONE 2010, 5, e9575. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Qi, X.; Chen, Z.; Shaw, L.; Cai, J.; Smith, L.H.; Grant, M.B.; Boulton, M.E. Targeting the IRE1A/XBP1 and ATF6 arms of the unfolded protein response enhances VEGF blockade to prevent retinal and choroidal neovascularization. Am. J. Pathol. 2013, 182, 1412–1424. [Google Scholar] [CrossRef] [PubMed]
- Romero-Ramirez, L.; Cao, H.; Regalado, M.P.; Kambham, N.; Siemann, D.; Kim, J.J.; Le, Q.T.; Koong, A.C. X box-binding protein 1 regulates angiogenesis in human pancreatic adenocarcinomas. Transl. Oncol. 2009, 2, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Ge, W.; Zhao, K.; Wang, X.; Li, H.; Yu, M.; He, M.; Xue, X.; Zhu, Y.; Zhang, C.; Cheng, Y.; et al. iASPP is an antioxidative factor and drives cancer growth and drug resistance by competing with Nrf2 for keap1 binding. Cancer Cell 2017, 32, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef] [PubMed]
- Bobrovnikova-Marjon, E.; Grigoriadou, C.; Pytel, D.; Zhang, F.; Ye, J.; Koumenis, C.; Cavener, D.; Diehl, J.A. Perk promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 2010, 29, 3881–3895. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Han, H.; Wu, L.; Pan, B.; Dong, B.; Yin, C.C.; Tian, Z.; Liu, X.; Yang, Y.; Zhang, H.; et al. iASPP facilitates tumor growth by promoting mTOR-dependent autophagy in human non-small-cell lung cancer. Cell Death Dis. 2017, 8, e3150. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Liu, Y.; Liu, H.; Chen, X.; Liu, M.; Che, H.; Guo, F.; Wang, C.; Zhang, D.; Wu, J.; et al. PERK silence inhibits glioma cell growth under low glucose stress by blockage of p-AKT and subsequent HK2’s mitochondria translocation. Sci. Rep. 2015, 5, 9065. [Google Scholar] [CrossRef] [PubMed]
- Romero-Ramirez, L.; Cao, H.; Nelson, D.; Hammond, E.; Lee, A.H.; Yoshida, H.; Mori, K.; Glimcher, L.H.; Denko, N.C.; Giaccia, A.J.; et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004, 64, 5943–5947. [Google Scholar] [CrossRef] [PubMed]
- Rouschop, K.M.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Spiotto, M.T.; Banh, A.; Papandreou, I.; Cao, H.; Galvez, M.G.; Gurtner, G.C.; Denko, N.C.; Le, Q.T.; Koong, A.C. Imaging the unfolded protein response in primary tumors reveals microenvironments with metabolic variations that predict tumor growth. Cancer Res. 2010, 70, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Dalton, L.E.; Clarke, H.J.; Knight, J.; Lawson, M.H.; Wason, J.; Lomas, D.A.; Howat, W.J.; Rintoul, R.C.; Rassl, D.M.; Marciniak, S.J. The endoplasmic reticulum stress marker chop predicts survival in malignant mesothelioma. Br. J. Cancer 2013, 108, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.P.; Barraclough, D.L.; Stewart, C.; Joyce, K.A.; Eccles, R.M.; Barraclough, R.; Rudland, P.S.; Sibson, D.R. Expression and splicing of the unfolded protein response gene XBP-1 are significantly associated with clinical outcome of endocrine-treated breast cancer. Int. J. Cancer 2008, 123, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Onda, M.; Nagai, H.; Nagahata, T.; Ogawa, K.; Emi, M. Upregulation and overexpression of human X-box binding protein 1 (hXBP-1) gene in primary breast cancers. Breast Cancer 2003, 10, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Yoshimatsu, K.; Watanabe, K.; Yokomizo, H.; Otani, T.; Matsumoto, A.; Osawa, G.; Onda, M.; Ogawa, K. Overexpression of human X-box binding protein 1 (XBP-1) in colorectal adenomas and adenocarcinomas. Anticancer Res. 2007, 27, 127–131. [Google Scholar] [PubMed]
- Kim, K.M.; Yu, T.K.; Chu, H.H.; Park, H.S.; Jang, K.Y.; Moon, W.S.; Kang, M.J.; Lee, D.G.; Kim, M.H.; Lee, J.H.; et al. Expression of er stress and autophagy-related molecules in human non-small cell lung cancer and premalignant lesions. Int. J. Cancer 2012, 131, E362–E370. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Kondoh, N.; Imazeki, N.; Tanaka, K.; Okada, T.; Mori, K.; Hada, A.; Arai, M.; Wakatsuki, T.; Matsubara, O.; et al. Activation of the ATF6, XBP1 and GRP78 genes in human hepatocellular carcinoma: A possible involvement of the er stress pathway in hepatocarcinogenesis. J. Hepatol. 2003, 38, 605–614. [Google Scholar] [CrossRef]
- Thorpe, J.A.; Schwarze, S.R. IRE1α controls cyclin A1 expression and promotes cell proliferation through XBP-1. Cell Stress Chaperones 2010, 15, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagratuni, T.; Wu, P.; Gonzalez de Castro, D.; Davenport, E.L.; Dickens, N.J.; Walker, B.A.; Boyd, K.; Johnson, D.C.; Gregory, W.; Morgan, G.J.; et al. XBP1S levels are implicated in the biology and outcome of myeloma mediating different clinical outcomes to thalidomide-based treatments. Blood 2010, 116, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, D.R.; Sukhdeo, K.; Protopopova, M.; Sinha, R.; Enos, M.; Carrasco, D.E.; Zheng, M.; Mani, M.; Henderson, J.; Pinkus, G.S.; et al. The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell 2007, 11, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schardt, J.A.; Weber, D.; Eyholzer, M.; Mueller, B.U.; Pabst, T. Activation of the unfolded protein response is associated with favorable prognosis in acute myeloid leukemia. Clin. Cancer Res. 2009, 15, 3834–3841. [Google Scholar] [CrossRef] [PubMed]
- Donze, O.; Jagus, R.; Koromilas, A.E.; Hershey, J.W.; Sonenberg, N. Abrogation of translation initiation factor eIF-2 phosphorylation causes malignant transformation of NIH 3T3 cells. EMBO J. 1995, 14, 3828–3834. [Google Scholar] [PubMed]
- Sequeira, S.J.; Ranganathan, A.C.; Adam, A.P.; Iglesias, B.V.; Farias, E.F.; Aguirre-Ghiso, J.A. Inhibition of proliferation by perk regulates mammary acinar morphogenesis and tumor formation. PLoS ONE 2007, 2, e615. [Google Scholar] [CrossRef] [PubMed]
- Ashktorab, H.; Green, W.; Finzi, G.; Sessa, F.; Nouraie, M.; Lee, E.L.; Morgano, A.; Moschetta, A.; Cattaneo, M.; Mariani-Costantini, R.; et al. Sel1l, an upr response protein, a potential marker of colonic cell transformation. Dig. Dis. Sci. 2012, 57, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M.; Fontanella, E.; Canton, C.; Delia, D.; Biunno, I. SEL1L affects human pancreatic cancer cell cycle and invasiveness through modulation of PTEN and genes related to cell-matrix interactions. Neoplasia 2005, 7, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Granelli, P.; Cattaneo, M.; Ferrero, S.; Bottiglieri, L.; Bosari, S.; Fichera, G.; Biunno, I. SEL1L and squamous cell carcinoma of the esophagus. Clin. Cancer Res. 2004, 10, 5857–5861. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chen, J.; Mai, B.; Amos, C.; Killary, A.M.; Sen, S.; Wei, C.; Frazier, M.L. A single-nucleotide polymorphism in tumor suppressor gene SEL1L as a predictive and prognostic marker for pancreatic ductal adenocarcinoma in caucasians. Mol. Carcinog. 2012, 51, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Mellai, M.; Cattaneo, M.; Storaci, A.M.; Annovazzi, L.; Cassoni, P.; Melcarne, A.; De Blasio, P.; Schiffer, D.; Biunno, I. SEL1L SNP rs12435998, a predictor of glioblastoma survival and response to radio-chemotherapy. Oncotarget 2015, 6, 12452–12467. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.H.; Mahon, P.C.; Oh, J.; Kelly, B.; Krishnamachary, B.; Pearson, M.; Chan, D.A.; Giaccia, A.J.; Semenza, G.L. OS-9 interacts with hypoxia-inducible factor 1α and prolyl hydroxylases to promote oxygen-dependent degradation of HIF-1α. Mol. Cell 2005, 17, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, K.; Konishi, H.; Arima, C.; Tomida, S.; Takeuchi, T.; Shimada, Y.; Yatabe, Y.; Mitsudomi, T.; Osada, H.; Takahashi, T. Novel metastasis-related gene cim functions in the regulation of multiple cellular stress-response pathways. Cancer Res. 2010, 70, 9949–9958. [Google Scholar] [CrossRef] [PubMed]
- Sha, H.; Sun, S.; Francisco, A.B.; Ehrhardt, N.; Xue, Z.; Liu, L.; Lawrence, P.; Mattijssen, F.; Guber, R.D.; Panhwar, M.S.; et al. The ER-associated degradation adaptor protein Sel1L regulates LPL secretion and lipid metabolism. Cell Metab. 2014, 20, 458–470. [Google Scholar] [CrossRef] [PubMed]
- DeLaBarre, B.; Christianson, J.C.; Kopito, R.R.; Brunger, A.T. Central pore residues mediate the p97/VCP activity required for erad. Mol. Cell 2006, 22, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, M.E.; Rico, C.; Tsoi, M.; Vivancos, M.; Filimon, S.; Paquet, M.; Boerboom, D. Pharmacological targeting of valosin containing protein (VCP) induces DNA damage and selectively kills canine lymphoma cells. BMC Cancer 2015, 15, 479. [Google Scholar] [CrossRef] [PubMed]
- Pasetto, M.; Antignani, A.; Ormanoglu, P.; Buehler, E.; Guha, R.; Pastan, I.; Martin, S.E.; FitzGerald, D.J. Whole-genome RNAI screen highlights components of the endoplasmic reticulum/Golgi as a source of resistance to immunotoxin-mediated cytotoxicity. Proc. Natl. Acad. Sci. USA 2015, 112, E1135–E1142. [Google Scholar] [CrossRef] [PubMed]
- Valle, C.W.; Min, T.; Bodas, M.; Mazur, S.; Begum, S.; Tang, D.; Vij, N. Critical role of VCP/p97 in the pathogenesis and progression of non-small cell lung carcinoma. PLoS ONE 2011, 6, e29073. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.P.; Beverly, L.J. Regulation of VCP/p97 demonstrates the critical balance between cell death and epithelial-mesenchymal transition (EMT) downstream of ER stress. Oncotarget 2015, 6, 17725–17737. [Google Scholar] [CrossRef] [PubMed]
- Cagnetta, A.; Caffa, I.; Acharya, C.; Soncini, D.; Acharya, P.; Adamia, S.; Pierri, I.; Bergamaschi, M.; Garuti, A.; Fraternali, G.; et al. APO866 increases antitumor activity of cyclosporin-a by inducing mitochondrial and endoplasmic reticulum stress in leukemia cells. Clin. Cancer Res. 2015, 21, 3934–3945. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, L.; Li, J.; Cheng, Y.; Chen, J.; Shen, M.; Zhang, S.; Wei, H. Insulin resistance contributes to multidrug resistance in HepG2 cells via activation of the PERK signaling pathway and upregulation of Bcl-2 and P-gp. Oncol. Rep. 2016, 35, 3018–3024. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, E.; Cao, Y.; Rodriguez, P.C. Endoplasmic reticulum stress regulates tumor growth and anti-tumor immunity: A promising opportunity for cancer immunotherapy. Cancer Immunol. Immunother. 2017, 66, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Riganti, C.; Kopecka, J.; Panada, E.; Barak, S.; Rubinstein, M. The role of C/EBP-β lip in multidrug resistance. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Salaroglio, I.C.; Panada, E.; Moiso, E.; Buondonno, I.; Provero, P.; Rubinstein, M.; Kopecka, J.; Riganti, C. Perk induces resistance to cell death elicited by endoplasmic reticulum stress and chemotherapy. Mol. Cancer 2017, 16, 91. [Google Scholar] [CrossRef] [PubMed]
- Dadey, D.Y.; Kapoor, V.; Khudanyan, A.; Urano, F.; Kim, A.H.; Thotala, D.; Hallahan, D.E. The ATF6 pathway of the ER stress response contributes to enhanced viability in glioblastoma. Oncotarget 2016, 7, 2080–2092. [Google Scholar] [CrossRef] [PubMed]
- Schewe, D.M.; Aguirre-Ghiso, J.A. ATF6α-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef] [PubMed]
- Hamada, A.; Miyano, H.; Watanabe, H.; Saito, H. Interaction of imatinib mesilate with human P-glycoprotein. J. Pharmacol. Exp. Ther. 2003, 307, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Kusio-Kobialka, M.; Podszywalow-Bartnicka, P.; Peidis, P.; Glodkowska-Mrowka, E.; Wolanin, K.; Leszak, G.; Seferynska, I.; Stoklosa, T.; Koromilas, A.E.; Piwocka, K. The PERK-eIF2α phosphorylation arm is a pro-survival pathway of BCR-ABL signaling and confers resistance to imatinib treatment in chronic myeloid leukemia cells. Cell Cycle 2012, 11, 4069–4078. [Google Scholar] [CrossRef] [PubMed]
- Maytin, E.V.; Habener, J.F. Transcription factors C/EBPα, C/EBPβ, and CHOP (Gadd153) expressed during the differentiation program of keratinocytes in vitro and in vivo. J. Investig. Dermatol. 1998, 110, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Meir, O.; Dvash, E.; Werman, A.; Rubinstein, M. C/EBP-β regulates endoplasmic reticulum stress-triggered cell death in mouse and human models. PLoS ONE 2010, 5, e9516. [Google Scholar] [CrossRef]
- Li, X.; Li, J.P.; Yuan, H.Y.; Gao, X.; Qu, X.J.; Xu, W.F.; Tang, W. Recent advances in P-glycoprotein-mediated multidrug resistance reversal mechanisms. Methods Find. Exp. Clin. Pharmacol. 2007, 29, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Globisch, C.; Pajeva, I.K.; Wiese, M. Structure-activity relationships of a series of tariquidar analogs as multidrug resistance modulators. Bioorg. Med. Chem. 2006, 14, 1588–1598. [Google Scholar] [CrossRef] [PubMed]
- Weidner, L.D.; Fung, K.L.; Kannan, P.; Moen, J.K.; Kumar, J.S.; Mulder, J.; Innis, R.B.; Gottesman, M.M.; Hall, M.D. Tariquidar is an inhibitor and not a substrate of human and mouse P-glycoprotein. Drug Metab. Dispos. 2016, 44, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.; Widemann, B.C.; Pastakia, D.; Chen, C.C.; Yang, S.X.; Cole, D.; Balis, F.M. Pharmacokinetic and pharmacodynamic study of tariquidar (XR9576), a P-glycoprotein inhibitor, in combination with doxorubicin, vinorelbine, or docetaxel in children and adolescents with refractory solid tumors. Cancer Chemother. Pharmacol. 2015, 76, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.R.; Chang, S.Y.; Hong, E.H.; Kwon, B.E.; Kim, H.M.; Kim, Y.J.; Lee, J.; Cho, H.J.; Cheon, J.H.; Ko, H.J. Elevated endoplasmic reticulum stress reinforced immunosuppression in the tumor microenvironment via myeloid-derived suppressor cells. Oncotarget 2014, 5, 12331–12345. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Lang, J.Y.; Hung, M.C.; Sengupta, K.; Banerjee, S.K.; Baksi, K.; Banerjee, D.K. Unfolded protein response is required in nu/nu mice microvasculature for treating breast tumor with tunicamycin. J. Biol. Chem. 2011, 286, 29127–29138. [Google Scholar] [CrossRef] [PubMed]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a novel perk kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Auf, G.; Jabouille, A.; Guerit, S.; Pineau, R.; Delugin, M.; Bouchecareilh, M.; Magnin, N.; Favereaux, A.; Maitre, M.; Gaiser, T.; et al. Inositol-requiring enzyme 1α is a key regulator of angiogenesis and invasion in malignant glioma. Proc. Natl. Acad. Sci. USA 2010, 107, 15553–15558. [Google Scholar] [CrossRef] [PubMed]
- Axten, J.M.; Romeril, S.P.; Shu, A.; Ralph, J.; Medina, J.R.; Feng, Y.; Li, W.H.; Grant, S.W.; Heerding, D.A.; Minthorn, E.; et al. Discovery of GSK2656157: An optimized perk inhibitor selected for preclinical development. ACS Med. Chem. Lett. 2013, 4, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Bi, M.; Naczki, C.; Koritzinsky, M.; Fels, D.; Blais, J.; Hu, N.; Harding, H.; Novoa, I.; Varia, M.; Raleigh, J.; et al. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 2005, 24, 3470–3481. [Google Scholar] [CrossRef] [PubMed]
- Chien, W.; Ding, L.W.; Sun, Q.Y.; Torres-Fernandez, L.A.; Tan, S.Z.; Xiao, J.; Lim, S.L.; Garg, M.; Lee, K.L.; Kitajima, S.; et al. Selective inhibition of unfolded protein response induces apoptosis in pancreatic cancer cells. Oncotarget 2014, 5, 4881–4894. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric inhibition of the IRE1α rnase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Iwakoshi, N.N.; Anderson, K.C.; Glimcher, L.H. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9946–9951. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.; Kebache, S.; Fazel, A.; Wong, H.N.; Jenna, S.; Emadali, A.; Lee, E.H.; Bergeron, J.J.; Kaufman, R.J.; Larose, L.; et al. Nck-dependent activation of extracellular signal-regulated kinase-1 and regulation of cell survival during endoplasmic reticulum stress. Mol. Biol. Cell 2004, 15, 4248–4260. [Google Scholar] [CrossRef] [PubMed]
- Niederreiter, L.; Fritz, T.M.; Adolph, T.E.; Krismer, A.M.; Offner, F.A.; Tschurtschenthaler, M.; Flak, M.B.; Hosomi, S.; Tomczak, M.F.; Kaneider, N.C.; et al. ER stress transcription factor Xbp1 suppresses intestinal tumorigenesis and directs intestinal stem cells. J. Exp. Med. 2013, 210, 2041–2056. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.H.; Ranatunga, S.; Kriss, C.L.; Cubitt, C.L.; Tao, J.; Pinilla-Ibarz, J.A.; Del Valle, J.R.; Hu, C.C. Inhibition of er stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J. Clin. Investig. 2014, 124, 2585–2598. [Google Scholar] [CrossRef] [PubMed]
- Cross, B.C.; Bond, P.J.; Sadowski, P.G.; Jha, B.K.; Zak, J.; Goodman, J.M.; Silverman, R.H.; Neubert, T.A.; Baxendale, I.R.; Ron, D.; et al. The molecular basis for selective inhibition of unconventional mrna splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. USA 2012, 109, E869–E878. [Google Scholar] [CrossRef] [PubMed]
- Mujcic, H.; Nagelkerke, A.; Rouschop, K.M.; Chung, S.; Chaudary, N.; Span, P.N.; Clarke, B.; Milosevic, M.; Sykes, J.; Hill, R.P.; et al. Hypoxic activation of the PERK/EIF2α arm of the unfolded protein response promotes metastasis through induction of LAMP3. Clin. Cancer Res. 2013, 19, 6126–6137. [Google Scholar] [CrossRef] [PubMed]
- Nagelkerke, A.; Bussink, J.; Mujcic, H.; Wouters, B.G.; Lehmann, S.; Sweep, F.C.; Span, P.N. Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast Cancer Res. 2013, 15, R2. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Denko, N.C.; Olson, M.; Van Melckebeke, H.; Lust, S.; Tam, A.; Solow-Cordero, D.E.; Bouley, D.M.; Offner, F.; Niwa, M.; et al. Identification of an ire1α endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 2011, 117, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Ri, M.; Tashiro, E.; Oikawa, D.; Shinjo, S.; Tokuda, M.; Yokouchi, Y.; Narita, T.; Masaki, A.; Ito, A.; Ding, J.; et al. Identification of toyocamycin, an agent cytotoxic for multiple myeloma cells, as a potent inhibitor of er stress-induced XBP1 mRNA splicing. Blood Cancer J. 2012, 2, e79. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, K.; Lucas, J.L.; Vuga, D.; Wang, X.; Brumm, D.; Stiles, C.; Kriebel, D.; Der-Sarkissian, A.; Krishnan, K.; Schweitzer, C.; et al. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J. Biol. Chem. 2011, 286, 12743–12755. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wei, L.; Fan, F.; Ji, S.; Zhang, S.; Geng, J.; Hong, L.; Fan, X.; Chen, Q.; Tian, J.; et al. Integration of hippo signalling and the unfolded protein response to restrain liver overgrowth and tumorigenesis. Nat. Commun. 2015, 6, 6239. [Google Scholar] [CrossRef] [PubMed]
- Morrow, K.; Hernandez, C.P.; Raber, P.; Del Valle, L.; Wilk, A.M.; Majumdar, S.; Wyczechowska, D.; Reiss, K.; Rodriguez, P.C. Anti-leukemic mechanisms of pegylated arginase I in acute lymphoblastic T-cell leukemia. Leukemia 2013, 27, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, S.T.; Carew, J.S.; Dunner, K., Jr.; Boise, L.H.; Chiao, P.J.; Huang, P.; Abbruzzese, J.L.; McConkey, D.J. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005, 65, 11510–11519. [Google Scholar] [CrossRef] [PubMed]
- Auner, H.W.; Moody, A.M.; Ward, T.H.; Kraus, M.; Milan, E.; May, P.; Chaidos, A.; Driessen, C.; Cenci, S.; Dazzi, F.; et al. Combined inhibition of p97 and the proteasome causes lethal disruption of the secretory apparatus in multiple myeloma cells. PLoS ONE 2013, 8, e74415. [Google Scholar] [CrossRef] [PubMed]
- Brem, G.J.; Mylonas, I.; Bruning, A. Eeyarestatin causes cervical cancer cell sensitization to bortezomib treatment by augmenting ER stress and CHOP expression. Gynecol. Oncol. 2013, 128, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, D.J.; Lefebvre, C.; Allan, K.; Brun, J.; Sanaei, C.A.; Baird, S.; Pearce, N.; Gronberg, S.; Wilson, B.; Prakesh, M.; et al. Virus-tumor interactome screen reveals er stress response can reprogram resistant cancers for oncolytic virus-triggered caspase-2 cell death. Cancer Cell 2011, 20, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M.; Baronchelli, S.; Schiffer, D.; Mellai, M.; Caldera, V.; Saccani, G.J.; Dalpra, L.; Daga, A.; Orlandi, R.; DeBlasio, P.; et al. Down-modulation of SEL1L, an unfolded protein response and endoplasmic reticulum-associated degradation protein, sensitizes glioma stem cells to the cytotoxic effect of valproic acid. J. Biol. Chem. 2014, 289, 2826–2838. [Google Scholar] [CrossRef] [PubMed]
- Zha, W.; Wang, G.; Xu, W.; Liu, X.; Wang, Y.; Zha, B.S.; Shi, J.; Zhao, Q.; Gerk, P.M.; Studer, E.; et al. Inhibition of P-glycoprotein by hiv protease inhibitors increases intracellular accumulation of berberine in murine and human macrophages. PLoS ONE 2013, 8, e54349. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, G.; Wei, H.; Sun, J.; Chen, J.; Xie, B.; Wang, B.; Gu, J.; Li, C.; Tian, B.; et al. The endoplasmic reticulum stress response is associated with insulin resistance-mediated drug resistance in HEPG2 cells. Neoplasma 2015, 62, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, G.; Mathur, A.; Mallade, P.; Gerlach, S.; Willis, J.; Datta, A.; Srivastav, S.; Abdel-Mageed, A.B.; Mondal, D. Nelfinavir targets multiple drug resistance mechanisms to increase the efficacy of doxorubicin in MCF-7/DOX breast cancer cells. Biochimie 2016, 124, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Asano, T.; Okubo, K.; Isono, M.; Asano, T. Nelfinavir and ritonavir kill bladder cancer cells synergistically by inducing endoplasmic reticulum stress. Oncol. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Leung-Hagesteijn, C.; Erdmann, N.; Cheung, G.; Keats, J.J.; Stewart, A.K.; Reece, D.E.; Chung, K.C.; Tiedemann, R.E. Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell 2013, 24, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.C.; Yang, J.S.; Lin, M.W.; Hsu, S.C.; Lin, J.J.; Lin, H.J.; Hsia, T.C.; Liao, C.L.; Yang, M.D.; Fan, M.J.; et al. Emodin has cytotoxic and protective effects in rat C6 glioma cells: Roles of Mdr1a and nuclear factor kB in cell survival. J. Pharmacol. Exp. Ther. 2009, 330, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Seres, M.; Ditte, P.; Breier, A.; Sulova, Z. Effect of thapsigargin on P-glycoprotein-negative and P-glycoprotein-positive L1210 mouse leukaemia cells. Gen. Physiol. Biophys. 2010, 29, 396–401. [Google Scholar] [CrossRef] [PubMed]
- El-Khattouti, A.; Sheehan, N.T.; Monico, J.; Drummond, H.A.; Haikel, Y.; Brodell, R.T.; Megahed, M.; Hassan, M. CD133(+) melanoma subpopulation acquired resistance to caffeic acid phenethyl ester-induced apoptosis is attributed to the elevated expression of ABCB5: Significance for melanoma treatment. Cancer Lett. 2015, 357, 83–104. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Liang, F.; Zhang, Z.; Liu, Y.; Sheng, H.; Xie, M. S1 kills MCF-7/ADR cells more than MCF-7 cells: A protective mechanism of endoplasmic reticulum stress. Biomed. Pharmacother. 2013, 67, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.L.; Chiang, P.C.; Guh, J.H. Tunicamycin induces resistance to camptothecin and etoposide in human hepatocellular carcinoma cells: Role of cell-cycle arrest and GRP78. Naunyn-Schmiedebergs Arch. Pharmacol. 2009, 380, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Kurtoglu, M.; Liu, H.; Wangpaichitr, M.; You, M.; Liu, X.; Savaraj, N.; Lampidis, T.J. 2-Deoxy-d-Glucose activates autophagy via endoplasmic reticulum stress rather than ATP depletion. Cancer Chemother. Pharmacol. 2011, 67, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, M.; Yoshimura, A.; Furukawa, T.; Sumizawa, T.; Nakazima, Y.; Akiyama, S. Glycosylation of P-glycoprotein in a multidrug-resistant kb cell line, and in the human tissues. Biochim. Biophys. Acta 1991, 1073, 309–315. [Google Scholar] [CrossRef]
- Zhang, Z.; Wu, J.Y.; Hait, W.N.; Yang, J.M. Regulation of the stability of P-glycoprotein by ubiquitination. Mol. Pharmacol. 2004, 66, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Kramer, R.; Weber, T.K.; Arceci, R.; Ramchurren, N.; Kastrinakis, W.V.; Steele, G., Jr.; Summerhayes, I.C. Inhibition of N-linked glycosylation of P-glycoprotein by tunicamycin results in a reduced multidrug resistance phenotype. Br. J. Cancer 1995, 71, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Pavlikova, L.; Seres, M.; Hano, M.; Bohacova, V.; Sevcikova, I.; Kyca, T.; Breier, A.; Sulova, Z. L1210 cells overexpressing ABCB1 drug transporters are resistant to inhibitors of the N- and O-glycosylation of proteins. Molecules 2017, 22, 1104. [Google Scholar] [CrossRef] [PubMed]
- Pavlikova, L.; Seres, M.; Imrichova, D.; Hano, M.; Rusnak, A.; Zamorova, M.; Katrlik, J.; Breier, A.; Sulova, Z. The expression of P-gp in leukemia cells is associated with cross-resistance to protein N-glycosylation inhibitor tunicamycin. Gen. Physiol. Biophys. 2016, 35, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Seres, M.; Cholujova, D.; Bubencikova, T.; Breier, A.; Sulova, Z. Tunicamycin depresses P-glycoprotein glycosylation without an effect on its membrane localization and drug efflux activity in L1210 cells. Int. J. Mol. Sci. 2011, 12, 7772–7784. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Jaffrey, S.R. Proteomic identification of protein ubiquitination events. Biotechnol. Genet. Eng. Rev. 2013, 29, 73–109. [Google Scholar] [CrossRef] [PubMed]
- Ronai, Z.A. Monoubiquitination in proteasomal degradation. Proc. Natl. Acad. Sci. USA 2016, 113, 8894–8896. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, A.; Chen, Z.J. Diversity of polyubiquitin chains. Dev. Cell 2009, 16, 485–486. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Duong, D.M.; Seyfried, N.T.; Cheng, D.; Xie, Y.; Robert, J.; Rush, J.; Hochstrasser, M.; Finley, D.; Peng, J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 2009, 137, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Chevet, E.; Hetz, C.; Samali, A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 2015, 5, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhu, W.; Wu, W. Micrornas change the landscape of cancer resistance. Methods Mol. Biol. 2018, 1699, 83–89. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |
Figure 1.
Biological function of P-glycoprotein (P-gp) in various tissues. (A) The blood-brain barrier separates blood circulating in the brain capillaries from the brain tissue. P-gp is one of the blood-brain barrier components and is localized on the luminal side of brain micro-capillary endothelial cells, where it prevents the penetration of xenobiotics through the endothelium to brain tissue; (B) P-gp promotes the excretion function of liver by transporting substances with limited solubility in water and their metabolites formed by enzymes of the first and second phase of liver detoxification machinery from hepatocytes into the bile; (C) P-gp in the proximal tubule cells of the nephrons secures elimination of water soluble substances via urine; (D) The gut blood barrier prevents the absorption of pathogens and toxins from diet [21,22]. P-gp is localized in the mucosal cells of the small intestine and promotes the integrity of the gut blood barrier by pumping harmful substances into the lumen of the intestines.
Figure 1.
Biological function of P-glycoprotein (P-gp) in various tissues. (A) The blood-brain barrier separates blood circulating in the brain capillaries from the brain tissue. P-gp is one of the blood-brain barrier components and is localized on the luminal side of brain micro-capillary endothelial cells, where it prevents the penetration of xenobiotics through the endothelium to brain tissue; (B) P-gp promotes the excretion function of liver by transporting substances with limited solubility in water and their metabolites formed by enzymes of the first and second phase of liver detoxification machinery from hepatocytes into the bile; (C) P-gp in the proximal tubule cells of the nephrons secures elimination of water soluble substances via urine; (D) The gut blood barrier prevents the absorption of pathogens and toxins from diet [21,22]. P-gp is localized in the mucosal cells of the small intestine and promotes the integrity of the gut blood barrier by pumping harmful substances into the lumen of the intestines.
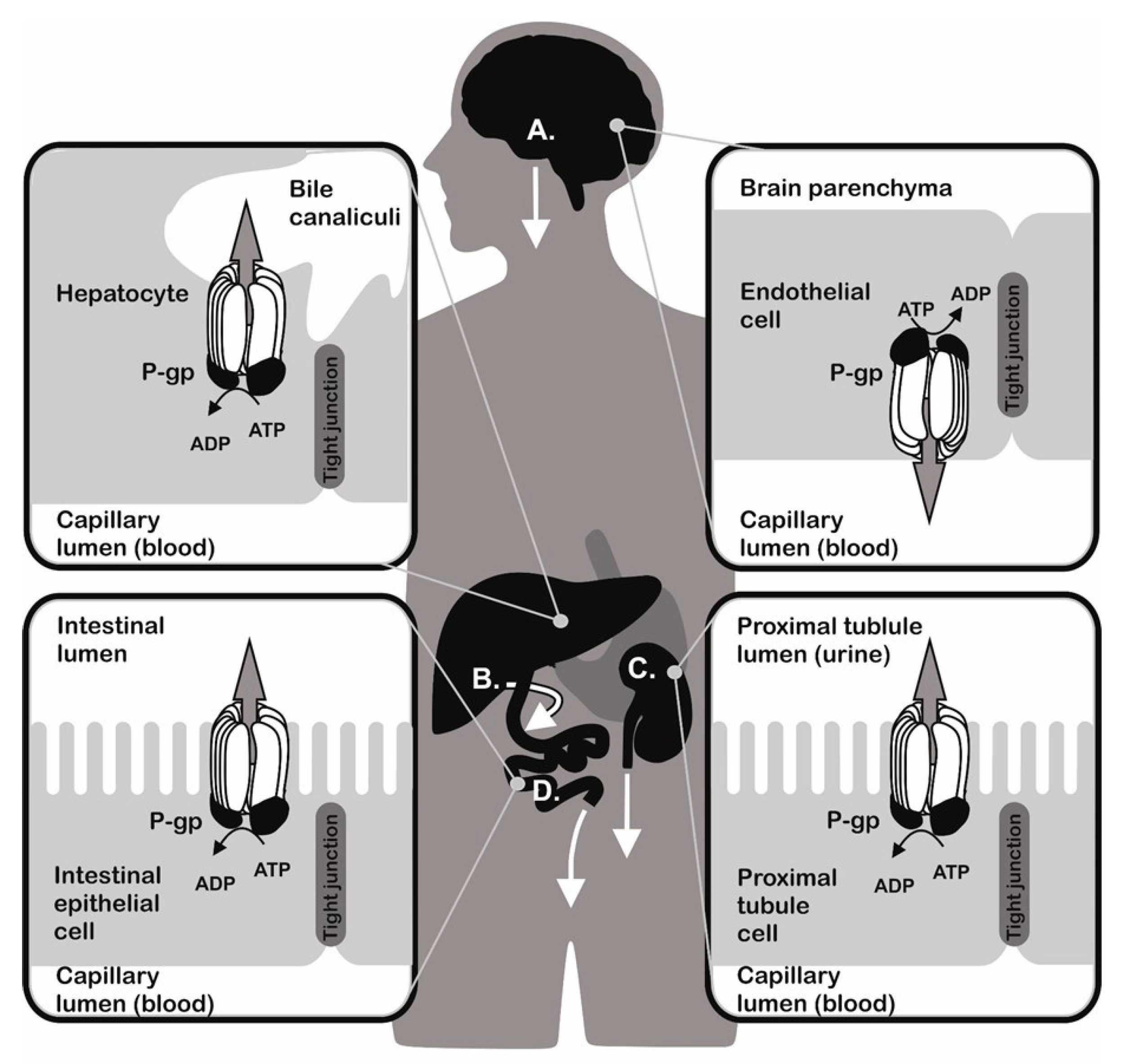
Figure 2.
N-glycosylation of protein in the endoplasmic reticulum (ER). (A) Synthesis of glycosylation core (Glc3Man9NAcGlc2) on dolichol attached to the ER membrane and oriented to the cytosol; (B) Relocation of the newly synthetized glycosylation core from the cytosolic to the luminal side of the ER by distributing flippase [58]; (C) Transfer of glycosylation core from dolichol phosphate to protein by oligosaccharyltransferase (EC 2.4.1.119) [62] and specific linkage of new glycoprotein with Ca2+-dependent lectins/chaperones of ER calnexin and calreticulin [63,64] due to its affinity for the oligosaccharide moiety labelled by a red square in A.
Figure 2.
N-glycosylation of protein in the endoplasmic reticulum (ER). (A) Synthesis of glycosylation core (Glc3Man9NAcGlc2) on dolichol attached to the ER membrane and oriented to the cytosol; (B) Relocation of the newly synthetized glycosylation core from the cytosolic to the luminal side of the ER by distributing flippase [58]; (C) Transfer of glycosylation core from dolichol phosphate to protein by oligosaccharyltransferase (EC 2.4.1.119) [62] and specific linkage of new glycoprotein with Ca2+-dependent lectins/chaperones of ER calnexin and calreticulin [63,64] due to its affinity for the oligosaccharide moiety labelled by a red square in A.

Figure 3.
The role of Endoplasmic Reticulum Stress (ERS) in tumour progression and development of multidrug resistance. The adaptation to ERS leads to deregulation of UPR, activation of pro survival pathways, suppression of apoptotic pathways and overexpression of P-glycoprotein (P-gp) in malignant cells.
Figure 3.
The role of Endoplasmic Reticulum Stress (ERS) in tumour progression and development of multidrug resistance. The adaptation to ERS leads to deregulation of UPR, activation of pro survival pathways, suppression of apoptotic pathways and overexpression of P-glycoprotein (P-gp) in malignant cells.
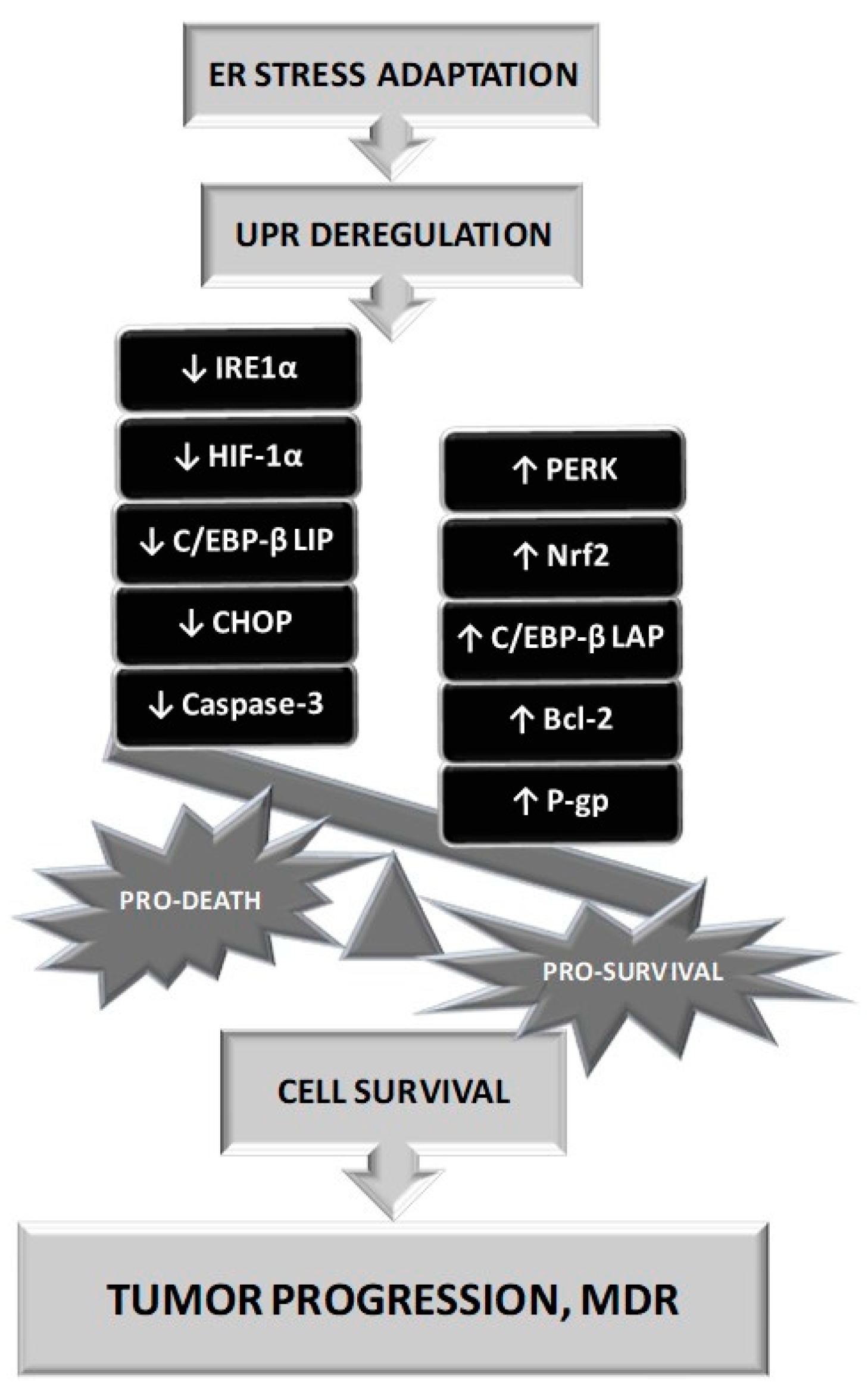
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hano, M.; Tomášová, L.; Šereš, M.; Pavlíková, L.; Breier, A.; Sulová, Z. Interplay between P-Glycoprotein Expression and Resistance to Endoplasmic Reticulum Stressors. Molecules 2018, 23, 337. https://doi.org/10.3390/molecules23020337
AMA Style
Hano M, Tomášová L, Šereš M, Pavlíková L, Breier A, Sulová Z. Interplay between P-Glycoprotein Expression and Resistance to Endoplasmic Reticulum Stressors. Molecules. 2018; 23(2):337. https://doi.org/10.3390/molecules23020337
Chicago/Turabian StyleHano, Milan, Lenka Tomášová, Mário Šereš, Lucia Pavlíková, Albert Breier, and Zdena Sulová. 2018. "Interplay between P-Glycoprotein Expression and Resistance to Endoplasmic Reticulum Stressors" Molecules 23, no. 2: 337. https://doi.org/10.3390/molecules23020337